Folha em branco:
Análise computacional do planejamento sintético: Passado e futuro
Adaptado de
Wang, Z., Zhang, W. e Liu, B. (2021), Computational Analysis of Synthetic Planning: Past and Future. Chin. J. Chem., 39: 3127-3143. https://doi.org/10.1002/cjoc.202100273
Publicado com a cortesia da Wiley.
O planejamento de síntese auxiliado por computador (CASP) pode desempenhar um papel significativo na organização e no aproveitamento do fluxo de novas reações químicas e regras de reação especializadas para o planejamento de uma síntese nova e altamente eficiente de produtos naturais e candidatos a medicamentos. Esta revisão descreve o progresso na análise computacional do planejamento sintético, desde o estágio inicial focado em programas baseados em regras até o aprendizado de máquina e sua capacidade combinada.
Introdução
Os químicos usam a análise retrosintética para projetar uma estratégia sintética para um composto-alvo. Em resumo, eles usam suas experiências na quebra de ligações químicas no composto-alvo e nos precursores subsequentes de forma iterativa.
Várias ferramentas padronizadas (por exemplo, CML, SMILES, SMARTS, InChl e ECFP) traduzem reações químicas e moléculas em informações legíveis por máquina. Algoritmos mais avançados (por exemplo, redes neurais, aprendizado por reforço) expandem o processamento de dados de reações químicas.
Esta análise abrange três categorias de CASP. Duas categorias usam a dedução lógica das intuições e experiências dos químicos: Algoritmos CASP baseados em regras codificadas manualmente ou em regras extraídas automaticamente. A terceira categoria de CASP usa Bancos de dados de reações químicas para treinamento de algoritmos de aprendizado de máquina (ML).
Estrutura geral do sistema CASP
Um sistema CASP típico tem quatro módulos. O banco de dados de modelos de reações armazena reações conhecidas com regras de quebra de ligações. O módulo retrosintético alinha as reações conhecidas no banco de dados de modelos com as estruturas das moléculas de entrada e fornece a correspondência mais próxima aos precursores disponíveis comercialmente de forma iterativa. O guia de árvore e o módulo de avaliação avaliam a adequação dos precursores candidatos às Rotas sintéticas. O banco de dados de compostos disponíveis comercialmente funciona como um ponto de parada para o módulo de retrosíntese.
Regras codificadas manualmente combinadas com um algoritmo lógico
Os sistemas CASP representativos incluem LHASA, SECS, IGOR, CHIRON e Chematica/SynthiaTM. Os sistemas LHASA e SECS CASP incluíam um módulo de comunicação: um bloco de notas com interface para que os químicos pudessem avaliar e selecionar a melhor rota da árvore sintética.
O IGOR (Intermediate Generation of Organic Reactions) não restringiu a análise retrosintética a regras heurísticas derivadas empiricamente. O IGOR inclui todas as moléculas que participam de uma reação, exige cálculos extensos e pode simular apenas transformações retrosintéticas simples.
O CHIRON pode decodificar a estereoquímica e a funcionalidade complexas, que podem ser correlacionadas a precursores enriquecidos com estereoquímica disponíveis comercialmente. Ele procura precursores com esqueletos, estereocentros e grupos funcionais estreitamente relacionados à molécula alvo.
O Chematica (agora chamado SynthiaTM) expandiu a Rede de Química Orgânica (NOC) para aproximadamente 10 milhões de compostos e adicionou manualmente informações de compatibilidade e contexto (por exemplo, condições canônicas, intolerância de grupos funcionais, regio e estereosseletividade de reações específicas) usando o método de codificação SMILES/SMART. Suas regras de reação codificadas manualmente aumentaram para mais de 100.000 em 2021. O Chematica/SynthiaTM incorporou uma função de pesquisa inteligente e funções de pontuação química que permitem resultados globalmente ideais (por exemplo, precursor quiral para síntese assimétrica).
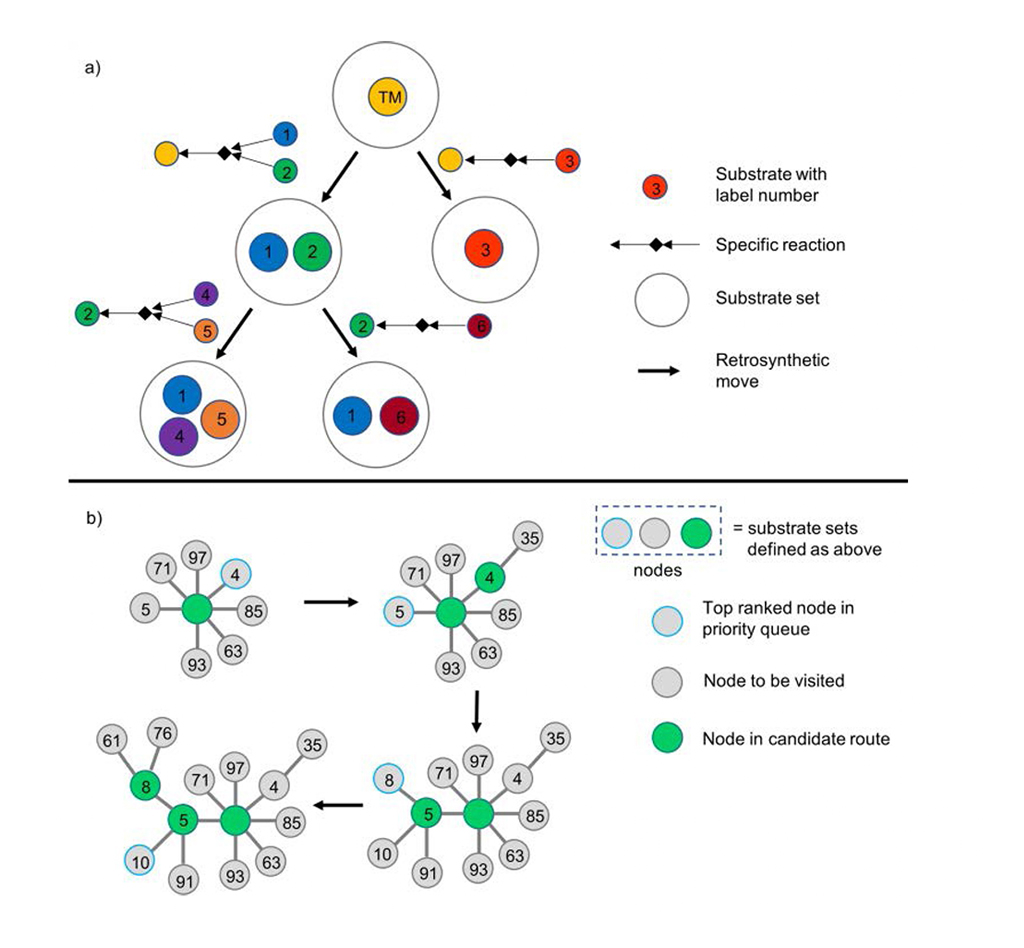
O Chematica/SynthiaTM apresenta a árvore sintética de forma dendrítica: cada nó denota a transformação retrossintética e seu conjunto de substratos associado (Fig. 1a). O Chematica/SynthiaTM acelera o processo analítico com uma fila de prioridades para os nós de pontuação mais baixa no algoritmo de pesquisa (Fig. 1b).
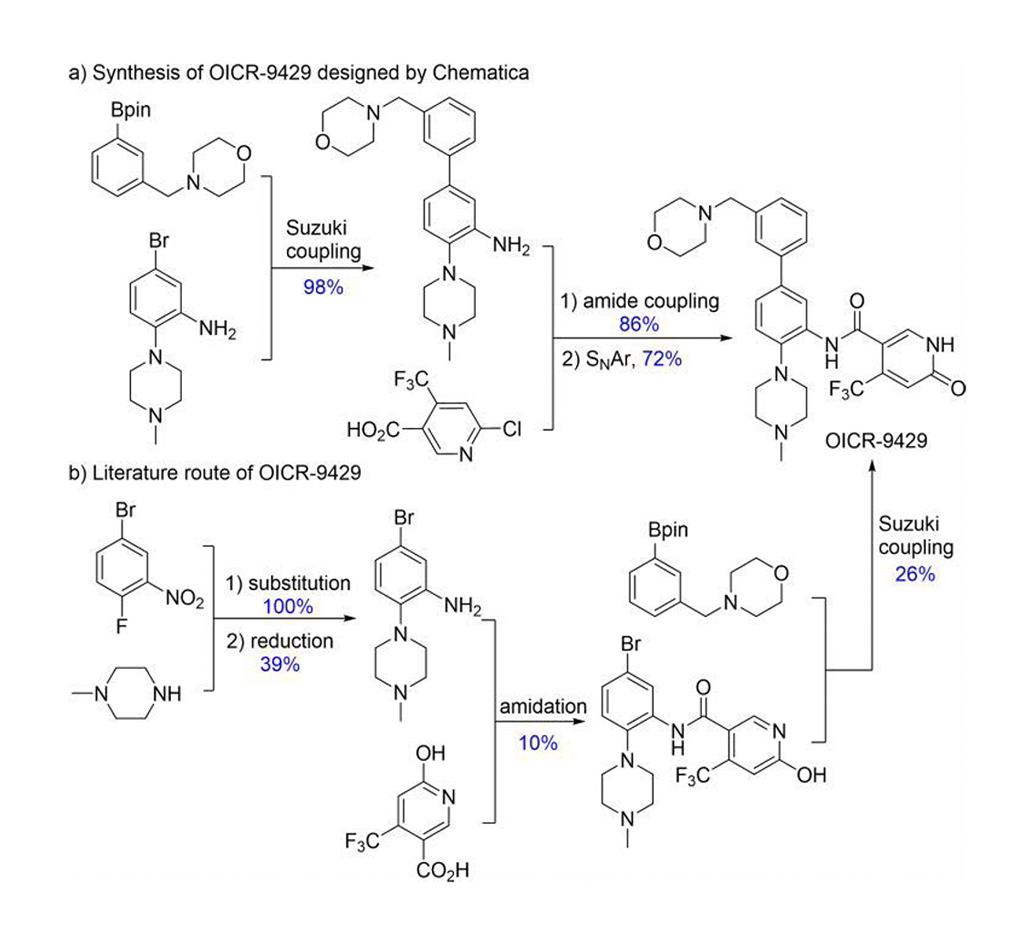
O Chematica/SynthiaTM inclui vários métodos de mecânica quântica e aprendizado de máquina (ML) para otimizar o algoritmo de busca, as funções de pontuação e as transformações estereosseletivas. O Chematica/SynthiaTM projetou rotas sintéticas para oito moléculas relacionadas a medicamentos e vários produtos naturais complexos. Suas sínteses foram realizadas experimentalmente. O programa SynthiaTM projetou uma rota sintética mais eficiente para o OICR-9429 (Fig. 2). A literatura relatou um rendimento de 1% do OICR-9429, mas a rota do SynthiaTM produziu 60%. Além disso, a rota sintética projetada pelo SynthiaTM simplificou sua purificação de quatro procedimentos cromatográficos para uma recristalização. Assim, Grzybowski e colegas de trabalho demonstram claramente que o Chematica/SynthiaTM pode resolver problemas complexos em química sintética.
A extração manual de modelos de reação pode ampliar as informações de contexto das reações químicas e aprimorar as análises retrosintéticas. A escolha entre a extração automática e a manual depende da descrição consistente das variáveis e das aplicações desejadas.
Regras extraídas automaticamente combinadas com um algoritmo lógico
A extração automática de novas reações químicas e modelos diariamente pode manter bancos de dados com eficiência, mas pode perder grupos funcionais e átomos adjacentes.
O SYNCHEM2 permite transformações sintéticas para trás e para frente com codificação alternativa. O RETROSYN abstrai o centro de reação e cria uma correlação atômica entre Produtos e Reagentes com um algoritmo especial de diferença de gráficos. O RETROSYN pesquisa e classifica o grau de correspondência com uma prioridade de alta a baixa, mas ignora a estereoquímica.
O KOSP (Kowledge-Base-Oriented System for Planejamento de síntese) extrai automaticamente os modelos de reação, incluindo a ativação de grupos/átomos dentro de três distâncias de ligação para preencher a Reaction Knowledge Base. A nova versão do KOSP permite a análise de retrosíntese regio e estereosseletiva e as atualizações expandiram o conteúdo da reação em 10 vezes.
O ChemPlanner, sucessor do ARChem, tem uma cooperação exclusiva com o American Chemical Abstracts Service e a Wiley para o SciFinder, um banco de dados altamente acessível de conteúdo de reações com curadoria científica. A nova versão do ChemPlanner permite a análise de retrosíntese regio e estereosseletiva.
O ICSYNTH representa seu banco de dados de conhecimento de reações na forma de gráficos. Os usuários podem incluir regras químicas internas do seu banco de dados confidencial de reações e adaptar o ICSYNTH para vários cenários de aplicação, selecionando e editando regras químicas.
O ASKCOS calcula a similaridade dos produtos da reação com a molécula alvo para desenvolver um plano retrosintético de forma gradual. Os módulos do ASKCOS incluem Retrossíntese em uma etapa, Planejamento interativo de caminhos, Construtor de árvores e Recomendação de contexto.
Regras extraídas automaticamente combinadas com algoritmo de aprendizado de máquina
Os algoritmos de ML são treinados com bancos de dados de reações químicas, incluindo reagentes. Os algoritmos de aprendizagem por reforço interagem continuamente com o ambiente, que lhes ensina a estratégia ideal por meio de uma abordagem de penalidade-recompensa.
O programa Bishop combina análise retrosintética baseada em regras e aprendizagem por reforço. A Rede de Reações Químicas compila os intermediários, conecta reagentes e produtos e tem um módulo de aprendizado por reforço para mapear caminhos de reação ideais e definidos de forma flexível com possíveis filtros para custo, Eficiência geral e/ou impactos ambientais.
O 3N-MCTS (algoritmo Monte Carlo Tree Search) usa as redes neurais artificiais treinadas por sequências digitais de Produtos e precursores relevantes da literatura. O sistema CASP baseado em ANN reorganiza as regras de reação específicas aprendidas, o que simplifica o processo de cálculo. Cada rodada do MCTS consiste em seleção, expansão, lançamento e atualização. São necessários aprimoramentos para prever estereosseletividades.
O modelo Seq2Seq com a tradução do SMILES (Simplified Molecular Input Line-Entry System)
pode processar um conjunto de dados maciço e simular uma reação com saída global ideal. O AutoSynRoute avalia as Vias sintéticas aplicando o algoritmo MCTS com funções de pontuação heurística inspiradas no Chematica/SynthiaTM. O RXN usa dois modelos retrosintéticos de ML treinados por dois bancos de dados. O RXN pode prever as condições de reação adequadas para a rota sintética proposta.
Conclusões
Vários programas CASP aplicam regras de reação heurísticas e regras de reação da literatura em seus algoritmos para química retrosintética com ou sem funções de pontuação e ML (por exemplo, Chematica/SynthiaTM). Outros programas CASP dependem de ML ou da combinação de ML com regras de reação heurísticas e/ou regras químicas baseadas na literatura. Esses algoritmos já forneceram Rotas sintéticas novas que melhoraram o rendimento de moléculas complicadas. Outras melhorias podem fornecer novas rotas sintéticas para compostos complexos com restrições adicionais, como menor custo, menor impacto ambiental e menos reagentes ou solventes perigosos.
