应用说明:
Synthia 合成实用性评分 (SAS)
在几分钟内评估数千个虚拟分子的合成实用性评分 (SAS)
用SYNTHIA®逆合成软件互联您的化学信息学工具
API 访问
API 访问(应用程序接口)适用于希望将其他化学信息学工具与SYNTHIA®互联以获得定制体验的组织。
优势包括
- 访问完整的逆合成或合成实用性评分 (SAS) API
- 并排查看数据,提高对分子选择的洞察力
- 使用多个数据源创建强大的可视化
- 为合成步骤上游的分子选择提供信息
- 使用 SAS API 在几分钟内分析数千条路线
利用合成实用性评分 (SAS) 的强大功能
区分 "易合成 "和 "难合成 "分子的能力是一项艰巨但却非常有用的任务,例如,在虚拟筛选管道中对化合物进行优先排序。通过将现代深度学习模型与我们著名的回溯合成规划软件收集的数据相结合,我们提供了SYNTHIA®合成实用性评分 (SAS) 服务,这是一种适用于高通量硅内化合物处理的工具。
目前,组合化学和生成模型被用于构建巨大的化合物数据集[1]。然而,利用这些方法获得的
许多分子的实际合成可能具有挑战性。为了解决这个问题,在药物发现过程中,合成可及性措施被用来尽早确定分子的可行性。
SYNTHIA®SAS API 服务提供了对这种 "分子复杂性 "的预测,即从市场上可买到的小砌块合成步骤的数量。支持 SAS 的机器学习模型已在使用SYNTHIA®逆向合成规划工具 [2]、[3]、[4] 算法获得的合成方案上进行了预训练。最后,我们的云托管产品通过了 ISO-27001 认证,每天可轻松处理数百万个分子,单次查询可处理多达上千个分子,从而使SYNTHIA®SAS 服务预测在药物设计过程中得到更广泛的应用。
SAS 模型的输入/输出
输入分子需要以广泛使用的 SMILES 文本格式[5]提供,API 端点支持批量请求。输入的 SMILES 由单个片段分子组成。返回的测量值(此处定义为合成实用性评分 (SAS))是一个浮点数,范围为 0-10,分配给每个相应的输入分子。返回的分数近似于使用市售合成砌块合成分子所需的步骤。最低分值(接近 0 的值)返回给预测容易制造(甚至可以在市场上买到)的化学物质。当模型预测需要更多的合成步骤才能获得所需的化合物时,返回的数值会更高。对于接近最大值(10)的分数,预测合成会非常复杂(反应步骤多),甚至不可行,例如,由于分子中存在奇特的结构基团。一般来说,分值越低,合成分子越容易。
如果请求中的某些分子无效(如高价、环不完整、芳香原子质子化不当、多片段),请求仍将被处理。此类条目的得分将为空,并在响应结构中返回相应的注释。
预测模型特征
SYNTHIA®SAS v1.0 基于包含图卷积神经网络(GCNN)的回归器。这种结构允许通过图结构而不是预先计算的分子描述符来学习每个分子的内部表示[6]。具体而言,该模型由键级有向信息传递神经网络(D-MPNN)和前馈神经网络(FNN)组成。
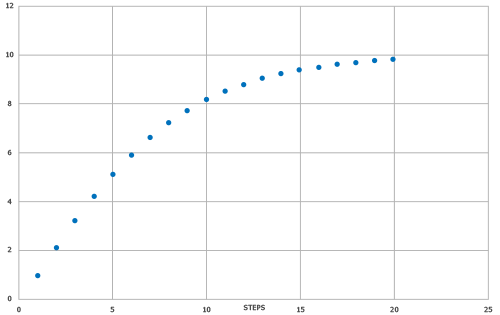
机器学习模型以SYNTHIA®自动逆合成模块的结果为目标值进行训练。为反映步骤的数量,使用了专门的归一化SYNTHIA®分数,例如,不惩罚非选择性反应、隐性保护策略、最小价格对分数的贡献,以及只使用小合成砌块作为SYNTHIA®搜索设置。此外,还应用了平滑函数,以更好地为高分建立梯度,目的是更好地解析难以合成的分子(另见图 1)。
用于训练机器学习模型的数据共有 33306 个分子。它由已知分子(ChEMBL 数据库)[8] 和组合生成的小分子(GDB)[9] 组成。训练/测试分割前的数据组成:
- GDB 子集:16081 个,包括:
- 含 1-7 个重原子(C、N、O、Cl、S)的化合物:7198
- 含 8-9 个重原子(C、N、O)的化合物:8883
- ChEMBL 子集:17225,包括:
- 随机选择的合成小化合物:15449
- 随机选择的天然产品衍生化合物:1776
机器学习模型的训练和评估需要将数据分成训练集和测试集(使用常见的 80/20 训练/测试分割)。此外,
,内部验证集以 9:1 的比例从训练集中提取,用于网络参数优化。
预测得分(SYNTHIA® SAS 模型)与基于SYNTHIA®分数的目标值相关,R2 = 0.726,MAE = 1.1497。带有拟合线的散点图和显示数据点密度/分布的方框图见图 2。

使用SYNTHIA®SAS 预测的结果是基于从数据集中检索到的关系(可能相当复杂,不容易捕捉)。通过SYNTHIA®SAS-API 查询新分子时应考虑到这一点。也就是说,与测试集无关的分子的得分可能会超出所谓的适用范围,因此相应的结果可能没有意义。这是数据驱动模型的典型局限性,不过,记住这种局限性对避免误读所获得的分数总是有好处的。
案例研究
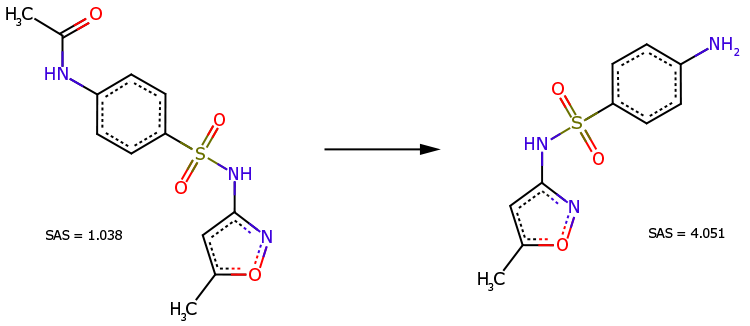
案例 1
磺胺甲噁唑的 N-乙酰基衍生物(图 3 左)是这种药物的直接前体(图 3 右)。尽管该衍生物的化学结构更为复杂,但它被认为更容易合成(SAS=1.038 比 SAS=4.051 小得多)。
案例 2
另一方面,肾上腺素的 N-Boc 衍生物(图 4 左)并不是肾上腺素的直接前体(图 4 右)。在典型的程序中,整个合成过程都不需要保护氨基基团。N-Boc 衍生物被正确地认为在合成可及性方面更为复杂(SAS=8.399 大于 SAS=7.631)。这与肾上腺素是其 N-Boc 衍生物的前体这一事实是一致的。

用户数据流
SYNTHIA®SAS 是一项云托管服务,每个客户都可通过 RESTful API 使用。它可横向扩展,并通过单一 API 入口点为所有客户提供高通量。最终用户需要提供 SMILES 格式的分子列表,SYNTHIA® SAS 会返回每个分子的得分(图 5)。该服务是无状态的,可根据需求进行扩展。

参考文献
1.Joshua Meyers, Benedek Fabian, Nathan Brown, De novo molecular design and generative models, Drug Discovery Today, 26,2021, 2707-2715.DOI
2.SYNTHIA®逆合成软件
3.Tomasz Klucznik, et al., Efficient Syntheses of Diverse, Medicinally Relevant Targets Planned by Computer and Executed in the Laboratory, Chem, 4,2018,
522-532.DOI
4.Mikulak-Klucznik, B., et al. Computational planning of the synthesis of complex natural products, Nature, 588,2020, 83-88.DOI
5.Daylight Chemical Information Systems, Inc.
6. Yang, K., et al. Analyzing Learned Molecular Representations for Property Prediction, Journal of Chemical Information and Modeling, 59,2019, 3370-3388.DOI
7.Chemprop 开源项目
8.ChEMBL 数据库
9.GDB 数据库
