Note d'application :
Score d'accessibilité synthétique (SAS) Synthia
Évaluer les scores d'accessibilité synthétique (SAS) de milliers de molécules virtuelles en quelques minutes.
Interconnectez vos outils de calcul de la chimie avec le Logiciel de rétrosynthèse SYNTHIA®.
Accès API
L'accès API (Application Programming Interface) est disponible pour les organisations qui souhaitent interconnecter d'autres outils de Cheminformatique avec SYNTHIA® pour une expérience personnalisée.
Les avantages sont les suivants
- Accès à la rétrosynthèse complète ou au Score d'accessibilité synthétique (SAS) API
- Visualisation des données côte à côte pour améliorer la compréhension de la sélection des molécules
- Création de visualisations robustes à l'aide de sources de données multiples
- Sélection des molécules en amont de l'étape de synthèse
- Analyse de milliers de voies en quelques minutes avec l'API SAS.
Exploiter la puissance du Score d'accessibilité synthétique (SAS)
La capacité à différencier les molécules "faciles à faire" des molécules "difficiles à faire" est une tâche difficile, mais largement utile, par exemple pour hiérarchiser les composés dans les pipelines de criblage virtuel. En combinant le modèle moderne d'apprentissage profond, et les données collectées avec notre célèbre logiciel de planification rétrosynthétique, nous fournissons le service SYNTHIA® Score d'accessibilité synthétique (SAS), un outil applicable au traitement des composés in-silico à haut débit.
À l'heure actuelle, la chimie combinatoire et la modélisation générative sont utilisées pour construire de gigantesques ensembles de données de composés [1]. Cependant, la synthèse effective de
nombreuses molécules obtenues avec ces méthodes peut s'avérer difficile. Pour résoudre ce problème, des mesures d'accessibilité synthétique sont utilisées pour déterminer la faisabilité des molécules le plus tôt possible dans le pipeline de découverte de médicaments.
Le service SYNTHIA® API de SAS permet de prédire cette "complexité moléculaire" en termes de nombre d'étapes de synthèse à partir de petits Building Blocks disponibles dans le commerce. Le modèle d'apprentissage automatique qui sous-tend SAS a été pré-entraîné sur des scénarios synthétiques obtenus avec les algorithmes de SYNTHIA® Retrosynthetic Planning Tool [2], [3], [4]. Enfin, notre produit hébergé dans le nuage et certifié ISO-27001 offre la possibilité de traiter facilement des millions de molécules par jour et jusqu'à un millier de molécules en une seule requête, ce qui permet d'utiliser plus couramment la prédiction de service de SYNTHIA® SAS dans le processus de conception de médicaments.
Entrée/sortie pour le modèle SAS
Les molécules d'entrée doivent être fournies dans le format texte SMILES largement utilisé [5] et le point de terminaison de l'API prend en charge les demandes par lots. Les SMILES d'entrée sont constitués d'un seul fragment de molécule. La mesure retournée, définie ici comme le Score d'accessibilité synthétique (SAS), est un nombre flottant unique compris entre 0 et 10, attribué à chaque molécule d'entrée correspondante. Le score retourné indique approximativement le nombre d'étapes nécessaires pour synthétiser la molécule en utilisant des Building Blocks disponibles dans le commerce. Les nombres les plus bas (valeurs proches de 0) sont renvoyés aux produits chimiques dont on prévoit qu'ils sont faciles à fabriquer (ou même qu'ils peuvent être disponibles dans le commerce). Les nombres les plus élevés sont renvoyés lorsque le modèle prévoit davantage d'étapes de synthèse pour obtenir le composé demandé. Pour les scores proches de la valeur maximale (10), il est prévu que la synthèse soit extrêmement complexe (nombreuses étapes de réaction) ou même irréalisable, par exemple en raison de motifs structurels exotiques dans la molécule. En général, plus le score est bas, plus il devrait être facile de synthétiser la molécule.
Si certaines des molécules de la demande ne sont pas valides (par exemple, hypervalentes, anneaux incomplets, protonation incorrecte des atomes aromatiques, multi-fragment), la demande sera tout de même traitée. Les scores pour ces entrées seront nuls et les commentaires appropriés seront renvoyés à côté dans la structure de la réponse.
Caractéristiques du modèle prédictif
SYNTHIA® SAS v1.0 est basé sur un régresseur qui inclut un réseau neuronal convolutionnel graphique (GCNN). Cette architecture permet d'apprendre une représentation interne de chaque molécule en opérant sur la structure de son graphe plutôt que sur des descripteurs moléculaires pré-calculés [6]. En particulier, le modèle se compose d'un réseau de neurones à passage de messages dirigés (D-MPNN) au niveau des liaisons, suivi d'un réseau de neurones feedforward (FNN). La mise en œuvre a été adaptée du projet open-source Chemprop [7].
Le modèle d'apprentissage automatique a été entraîné en utilisant les résultats du module de Rétrosynthèse automatique SYNTHIA® comme valeur cible. Un score SYNTHIA® spécialisé et normalisé a été utilisé pour refléter le nombre d'étapes, par exemple, ne pas pénaliser les réactions non sélectives, la stratégie des protections implicites, la contribution minimale du prix au score, et seuls les petits Building Blocks ont été utilisés comme paramètres de recherche SYNTHIA®. En outre, une fonction de lissage a été appliquée pour mieux construire le gradient pour les scores élevés, visant à une meilleure résolution des molécules difficiles à synthétiser (voir également la figure 1).
Les données utilisées pour l'entraînement des modèles d'apprentissage automatique comprennent 33306 molécules au total. Elles se composent de molécules connues (base de données ChEMBL) [8] et de petites molécules générées de manière combinatoire (GDB) [9]. La composition des données avant la séparation formation/test est la suivante :
- sous-ensemble GDB : 16081, dont :
- composés avec 1-7 atomes lourds (C, N, O, Cl, S) : 7198
- composés avec 8-9 atomes lourds (C, N, O) : 8883
- sous-ensemble ChEMBL : 17225, dont :
- petits composés synthétiques sélectionnés au hasard : 15449
- composés dérivés de produits naturels sélectionnés au hasard : 1776
La formation et l'évaluation du modèle d'apprentissage automatique ont nécessité la division des données en ensembles de formation et de test (une division commune 80/20 formation/test a été utilisée). En outre,
l'ensemble de validation interne a été extrait de l'ensemble de formation dans un rapport de 9:1 et a été utilisé pour l'optimisation des paramètres du réseau.
Le score prédit (modèle SYNTHIA® SAS) est en corrélation avec la valeur cible basée sur les scores SYNTHIA® avec R2 = 0,726 et MAE = 1,1497. Le diagramme de dispersion avec la ligne ajustée et le diagramme en boîte montrant la densité/la distribution des points de données sont présentés à la figure 2.

Les résultats prédits avec SYNTHIA® SAS sont basés sur des relations extraites d'ensembles de données (éventuellement assez complexes et difficiles à saisir). Il convient d'en tenir compte lorsque des molécules nouvelles sont interrogées via SYNTHIA® SAS-API. En effet, les scores des molécules qui ne sont pas liées à l'ensemble de test peuvent sortir du domaine dit d'applicabilité, et les résultats correspondants peuvent donc ne pas être significatifs. Il s'agit d'une limitation typique des modèles guidés par les données, mais il est toujours bon de s'en souvenir afin d'éviter une mauvaise interprétation des scores obtenus.
Études de cas
Cas 1
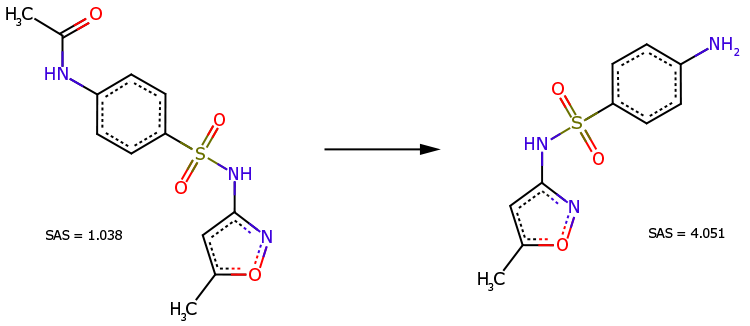
Le dérivé N-acétyle du sulfaméthoxazole (Fig. 3, à gauche) est un précurseur direct de ce médicament (Fig. 3, à droite). Malgré sa structure chimique plus complexe, le dérivé est reconnu comme étant plus facile à synthétiser (SAS=1,038 est beaucoup plus petit que SAS=4,051).
Cas 2
En revanche, le dérivé N-Boc de l'adrénaline (Fig. 4, à gauche) n'est pas un précurseur direct de l'adrénaline (Fig. 4, à droite). Dans la procédure typique, il n'est pas nécessaire de protéger le groupe amino tout au long de la voie de synthèse. Le dérivé N-Boc est correctement reconnu comme étant plus complexe en termes d'accessibilité synthétique (SAS=8,399 est supérieur à SAS = 7,631). Ceci est en accord avec le fait que l'adrénaline est un précurseur de son dérivé N-Boc.

Flux de données utilisateur
SYNTHIA® SAS est un service hébergé dans le cloud, disponible pour chaque client via une API RESTful. Il est évolutif horizontalement et offre un Haut débit via un point d'entrée API unique pour tous les clients. L'utilisateur final doit fournir une liste de molécules au format SMILES et SYNTHIA® SAS renvoie un score pour chacune d'entre elles (figure 5). Le service est sans état et conçu pour s'adapter à la demande.

Références
1. Joshua Meyers, Benedek Fabian, Nathan Brown, De novo molecular design and generative models, Découverte de médicaments Today, 26, 2021, 2707-2715. DOI
2.Logiciel de rétrosynthèse SYNTHIA®
3.Tomasz Klucznik, et al, Efficient Syntheses of Diverse, Medicinally Relevant Targets Planned by Computer and Executed in the Laboratory, Chem, 4, 2018,
522-532.DOI
4. Mikulak-Klucznik, B., et al. Planification computationnelle de la synthèse de produits naturels complexes, Nature, 588, 2020, 83-88. DOI
5.Daylight Chemical Information Systems, Inc.
6. Yang, K., et al. Analyzing Learned Molecular Representations for Property Prediction, Journal of chemical information and modeling, 59, 2019, 3370-3388. DOI
7.Projet open-source Chemprop
8.Base de données ChEMBL
9.Base de données GDB
