アプリケーションノート
Synthia 合成アクセシビリティスコア (SAS)
数千の仮想分子の合成アクセシビリティスコア(SAS)を数分で評価します。
SYNTHIA逆合成解析ソフトウェアとケムインフォマティクスツールの相互接続
APIアクセス
APIアクセス(Application Programming Interface)は、他のケムインフォマティクス・ツールとSYNTHIA®を相互接続して、カスタマイズされたエクスペリエンスを得たい組織向けに用意されています。
以下のような利点があります:
- 完全なレトロシンセシスまたは合成アクセシビリティスコア(SAS)APIへのアクセス
- データを並べて表示し、分子選択に関する洞察を向上
- 複数のデータソースを使用して堅牢なビジュアライゼーションを作成
- 合成ステップの上流で分子選択に情報を提供
- SAS APIを使用して数千の経路を数分で分析
合成アクセシビリティスコア(SAS)のパワーを活用する
作りやすい」分子と「作りにくい」分子を区別する能力は、バーチャル・スクリーニング・パイプラインにおける化合物の優先順位付けなど、難しいが広く有用なタスクである。最新のディープラーニングモデルと、当社の定評ある逆合成計画ソフトウェアで収集したデータを組み合わせることで、ハイスループットのインシリコ化合物処理に適用可能なツールであるSYNTHIA®合成アクセシビリティスコア(SAS)サービスを提供します。
現在、コンビナトリアルケミストリーとジェネレイティブモデリングは、巨大な化合物データセットの構築に用いられている[1]。しかし、このような手法で得られた
多数の分子を実際に合成することは困難である。この問題に対処するため、合成アクセシビリティ測定法は、医薬品探索パイプラインのできるだけ早い段階で分子の実現可能性を判断するために使用される。
SYNTHIA®SAS APIサービスは、市販されている小さなビルディングブロックから、合成ステップ数という観点から、このような「分子の複雑さ」の予測を提供します。SASを支える機械学習モデルは、SYNTHIA® Retrosynthetic Planning Tool [2], [3], [4]のアルゴリズムで得られた合成シナリオで事前にトレーニングされています。最後に、ISO-27001認証を取得した当社のクラウド製品は、毎日数百万個の分子を簡単に処理でき、1回のクエリで最大1,000個の分子を処理できるため、SYNTHIA® SASのサービス予測が創薬プロセスでより一般的に使用できるようになります。
SASモデルの入出力
入力分子は広く使用されているSMILESテキスト形式[5]で提供される必要があり、APIエンドポイントはバッチリクエストをサポートしている。入力SMILESは単一フラグメント分子から構成される。返される指標は、ここでは合成アクセシビリティスコア(SAS)と定義され、対応する入力分子ごとに割り当てられた0から10の範囲の浮動小数点数である。返されるスコアは、市販のビルディングブロックを使って分子を合成するのにかかるステップ数を概算したものである。最も低い数値(0に近い値)は、製造が簡単であると予測される(あるいは市販されている)化学物質に返されます。より高い数値は、モデルが要求された化合物を得るために、より多くの合成ステップを予測した場合に返される。最大値(10)に近いスコアでは、合成が非常に複雑(反応ステップが多い)か、分子内にエキゾチックな構造モチーフがあるなどの理由で実現不可能と予測される。一般的に、スコアが低いほどその分子を合成しやすいはずである。
リクエスト中の分子が無効な場合(例えば、超原子価、不完全な環、芳香族原子の不適切なプロトン化、マルチフラグメント)もリクエストは処理される。そのようなエントリーのスコアはNULLとなり、適切なコメントがレスポンス構造で一緒に返される。
予測モデルの特徴
SYNTHIA®SAS v1.0は、グラフ畳み込みニューラルネットワーク(GCNN)を含む回帰器に基づいています。このようなアーキテクチャは、事前に計算された分子記述子ではなく、グラフ構造を操作することによって、各分子の内部表現を学習することを可能にします[6]。特に、このモデルはボンドレベルの有向メッセージパッシングニューラルネットワーク(D-MPNN)とフィードフォワードニューラルネットワーク(FNN)から構成されている。
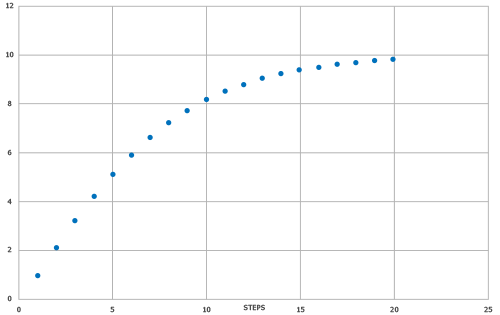
機械学習モデルは、SYNTHIA®自動Retrosynthesisモジュールの結果を目標値として使用して学習されました。例えば、非選択的反応にペナルティを課さない、暗黙の保護戦略、スコアへの最小限の価格寄与、小さなビルディングブロックのみをSYNTHIA®検索設定として使用した。さらに、合成が困難な分子の解像度を向上させることを目的として、高スコアに対してより良い勾配を構築するために平滑化関数を適用した(図1も参照)。
機械学習モデルの学習に使用するデータは、全部で33306分子である。これは既知の分子(ChEMBLデータベース)[8]とコンビナトリカルに生成された低分子分子(GDB)[9]から構成されている。train/test分割前のデータ構成:
- GDBサブセット:16081, 以下を含む:
- 1-7個の重原子(C, N, O, Cl, S)を持つ化合物: 7198
- 8-9個の重原子(C, N, O)を持つ化合物:8883
- ChEMBLサブセット:17225, 以下を含む:
- ランダムに選択した合成低分子化合物:15449
- ランダムに選択された天然物由来の化合物:1776
機械学習モデルの訓練と評価には、データを訓練セットとテストセットに分割する必要があった(一般的な80/20の訓練/テスト分割を使用)。さらに、
内部検証セットをトレーニングセットから9:1の割合で抽出し、ネットワークパラメータの最適化に使用。
予測スコア(SYNTHIA® SASモデル)は、SYNTHIA®スコアに基づく目標値と相関し、R2 = 0.726、MAE = 1.1497。フィットした直線を用いた散布図と、データ点の密度/分布を示す箱ひげ図を図2に示す。

SYNTHIA®SASで予測される結果は、データセットから検索された関係に基づいています(おそらく、非常に複雑で、一筋縄ではいかないでしょう)。新規分子をSYNTHIA® SAS-API で検索する場合は、この点を考慮する必要があります。すなわち、テストセットに関連しない分子のスコアは、いわゆる適用領域から外れる可能性があり、対応する結果は意味をなさないかもしれません。これはデータ駆動型モデルの典型的な制限であるが、得られたスコアの誤った解釈を避けるために、このような制限を常に覚えておくことは良いことである。
ケーススタディ
事例1
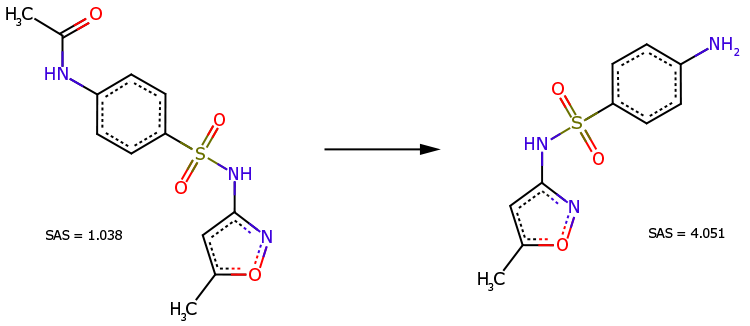
スルファメトキサゾールのN-アセチル誘導体(図3、左)は、この薬物の直接的な前駆体である(図3、右)。より複雑な化学構造にもかかわらず、この誘導体は合成が容易であると認識されている(SAS=1.038はSAS=4.051よりはるかに小さい)。
ケース2
一方、アドレナリンのN-Boc誘導体(図4、左)は、アドレナリン(図4、右)の直接的な前駆体ではない。一般的な手順では、合成経路を通してアミノ基を保護する必要はない。N-Boc誘導体は、合成アクセシビリティの点で、より複雑であると正しく認識される(SAS=8.399はSAS=7.631より大きい)。これは、アドレナリンがそのN-Boc誘導体の前駆体であるという事実と一致している。

ユーザーデータフロー
SYNTHIA®SASはクラウドホスティングサービスであり、RESTful APIを介して各顧客が利用できます。水平方向に拡張可能で、すべてのお客様に単一のAPIエントリーポイントを介してハイスループットを提供します。エンドユーザーは分子のリストをSMILESフォーマットで提供する必要があり、SYNTHIA® SASはそれぞれの分子のスコアを返す(図5)。このサービスはステートレスで、需要に応じて拡張できるように設計されている。

参考文献
1.Joshua Meyers, Benedek Fabian, Nathan Brown, De novo molecular design and generative models, Drug Discovery Today, 26,2021, 2707-2715.DOI
2.SYNTHIA®逆合成解析ソフトウェア
3.Tomasz Klucznik, et al., Computer Planned and Executed in the Laboratory, Chem, 4,2018,
522-532.DOI
4.Mikulak-Klucznik, B., et al. Computational planning of the synthesis of complex natural products, Nature, 588,2020, 83-88.DOI
5.Daylight Chemical Information Systems, Inc.
6. Yang, K., et al. Analyzing Learned Molecular Representations for Property Prediction, Journal of chemical information and modeling, 59,2019, 3370-3388.DOI
7.Chemprop オープンソースプロジェクト
8.ChEMBL データベース
9.GDB データベース
