애플리케이션 노트:
신시아 합성 접근성 점수 (SAS)
수천 개의 가상 분자에 대한 합성 접근성 점수(SAS)를 몇 분 안에 평가합니다.
케미포매틱스 툴과 SYNTHIA® 역합성 소프트웨어의 상호 연결
API 액세스
API 액세스(응용 프로그래밍 인터페이스)는 맞춤형 경험을 위해 다른 Cheminformatics 도구를 SYNTHIA®와 상호 연결하고자 하는 조직에서 사용할 수 있습니다.
혜택은 다음과 같습니다:
- 전체 재합성 또는 합성 접근성 점수(SAS) API 액세스
- 데이터를 나란히 확인하여 분자 선택에 대한 통찰력 향상
- 여러 데이터 소스를 사용하여 강력한 시각화 생성
- 합성 단계부터 분자 선택 정보를 업스트림으로 제공
- SAS API로 수천 개의 경로를 몇 분 안에 분석할 수 있습니다.
합성 접근성 점수(SAS)의 강력한 기능 활용하기
'만들기 쉬운' 분자와 '만들기 어려운' 분자를 구별하는 능력은 가상 스크리닝 파이프라인에서 화합물의 우선순위를 정하는 등 어렵지만 널리 유용한 작업입니다. 당사는 최신 딥러닝 모델과 수집된 데이터를 당사의 유명한 역합성 계획 소프트웨어와 결합하여 고처리량 인실리코 화합물 처리에 적용할 수 있는 도구인 SYNTHIA® 합성 접근성 점수(SAS) 서비스를 제공합니다.
현재 조합 화학 및 생성 모델링은 거대한 화합물 데이터 세트를 구축하는 데 사용됩니다[1]. 그러나 이러한 방법으로 얻은
많은 분자를 실제로 합성하는 것은 어려울 수 있습니다. 이 문제를 해결하기 위해 합성 접근성 측정은 약물 발견 파이프라인에서 가능한 한 빨리 분자의 실현 가능성을 결정하는 데 사용됩니다.
SYNTHIA® SAS API 서비스는 이러한 '분자 복잡성'을 상용화된 작은 빌딩 블록으로부터 합성 단계 수에 대한 예측을 제공합니다. SAS의 기반이 되는 머신러닝 모델은 SYNTHIA® Retrosynthetic Planning Tool [2], [3], [4]의 알고리즘으로 얻은 합성 시나리오에 대해 사전 학습을 거쳤습니다. 마지막으로, 클라우드 호스팅 및 ISO-27001 인증을 받은 이 제품은 단일 쿼리로 매일 수백만 개, 최대 수천 개의 분자를 쉽게 처리할 수 있는 기능을 제공하므로, 약물 설계 프로세스에서 SYNTHIA® SAS 서비스 예측을 보다 일반적으로 사용할 수 있습니다.
SAS 모델의 입력/출력
입력 분자는 널리 사용되는 SMILES 텍스트 형식[5]으로 제공되어야 하며 API 엔드포인트는 일괄 요청을 지원합니다. 입력 SMILES는 단일 조각 분자로 구성됩니다. 반환되는 측정값은 합성 접근성 점수(SAS)로 정의되며, 각 해당 입력 분자에 대해 할당된 0-10 범위의 단일 부동 소수점 숫자입니다. 반환된 점수는 상용 빌딩 블록을 사용하여 분자를 합성하는 데 걸리는 단계 수를 대략적으로 나타냅니다. 가장 낮은 숫자(0에 가까운 값)는 쉽게 만들 수 있을 것으로 예상되는(또는 상업적으로 이용 가능한) 화학 물질에 대해 반환됩니다. 모델이 요청된 화합물을 얻기 위해 더 많은 합성 단계를 예측할수록 높은 수치가 반환됩니다. 최대값(10)에 가까운 점수의 경우, 합성이 매우 복잡하거나(반응 단계가 많음) 분자의 구조적 모티프가 특이하여 합성이 불가능할 것으로 예측됩니다(예: 분자의 구조적 모티프가 특이함). 일반적으로 점수가 낮을수록 분자의 합성이 더 쉬울 것입니다.
요청된 분자 중 일부가 유효하지 않은 경우(예: 과원자, 불완전한 고리, 방향족 원자의 부적절한 양성자화, 다중 조각)에도 요청은 처리될 수 있습니다. 이러한 항목에 대한 점수는 0이 되며 응답 구조에 적절한 주석이 함께 반환됩니다.
예측 모델 특성
SYNTHIA® SAS v1.0은 그래프 합성곱 신경망(GCNN)을 포함하는 회귀 분석에 기반합니다. 이러한 아키텍처는 미리 계산된 분자 설명자가 아닌 그래프 구조에서 작동하여 각 분자의 내부 표현을 학습할 수 있습니다[6]. 특히 이 모델은 결합 수준 직접 메시지 전달 신경망(D-MPNN)과 피드포워드 신경망(FNN)으로 구성되며, 구현은 Chemprop 오픈 소스 프로젝트 [7]를 채택했습니다.
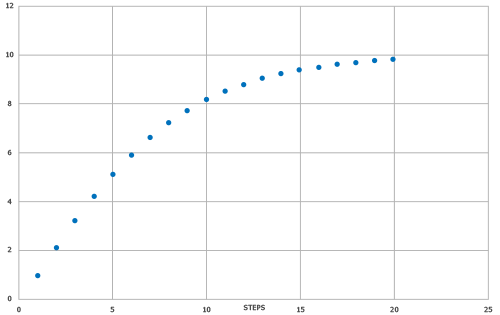
머신러닝 모델은 SYNTHIA® 자동 역합성 모듈 결과를 목표 값으로 사용하여 학습했습니다. 비선택적 반응에 불이익을 주지 않고, 암묵적 보호 전략, 점수에 대한 가격 기여도를 최소화하는 등 특수화되고 정규화된 SYNTHIA® 점수를 반영하기 위해 단계 수를 사용했으며, 작은 빌딩 블록만 SYNTHIA® 검색 설정으로 사용했습니다. 또한 합성하기 어려운 분자의 해상도를 높이기 위해 스무딩 기능을 적용하여 높은 점수에 대한 빌드 그라데이션을 개선했습니다(그림 1 참조).
머신러닝 모델 학습에 사용되는 데이터는 총 33306개의 분자로 구성되어 있습니다. 이는 알려진 분자(ChEMBL 데이터베이스)[8]와 조합적으로 생성된 작은 분자(GDB)[9]로 구성되어 있습니다. 훈련/테스트 분할 전 데이터 구성:
- GDB 하위 집합: 16081, 포함:
- 1~7개의 중원자(C, N, O, Cl, S)를 가진 화합물: 7198
- 8~9개의 중원자(C, N, O)를 가진 화합물: 8883
- ChEMBL 하위 집합: 17225, 포함:
- 무작위로 선택된 작은 합성 화합물: 15449
- 무작위로 선택된 천연 제품 유래 화합물: 1776
머신 러닝 모델의 훈련 및 평가에는 데이터를 훈련 및 테스트 세트로 분할해야 했습니다(일반적인 80/20 훈련/테스트 분할이 사용됨). 또한
내부 검증 세트는 훈련 세트에서 9:1 비율로 추출하여 네트워크 파라미터 최적화에 사용했습니다.
예측 점수(SYNTHIA® SAS 모델)는 R2 = 0.726, MAE = 1.1497의 SYNTHIA® 점수를 기반으로 목표 값과 상관관계가 있습니다. 그림 2에는 데이터 포인트의 밀도/분포를 보여주는 적합 선과 박스 플롯이 포함된 산점도가 나와 있습니다.

SYNTHIA® SAS로 예측한 결과는 데이터 세트에서 검색된 관계를 기반으로 합니다(매우 복잡하고 캡처하기 쉽지 않을 수 있음). SYNTHIA® SAS-API를 통해 신규 분자를 쿼리할 때는 이 점을 고려해야 합니다. 즉, 테스트 세트와 관련이 없는 분자에 대한 점수는 소위 적용 가능성 영역에서 벗어날 수 있으므로 해당 결과가 의미가 없을 수 있습니다. 이는 데이터 기반 모델의 일반적인 제한 사항이지만, 얻은 점수를 잘못 해석하지 않도록 항상 이러한 제한 사항을 기억해 두는 것이 좋습니다.
사례 연구
사례 1
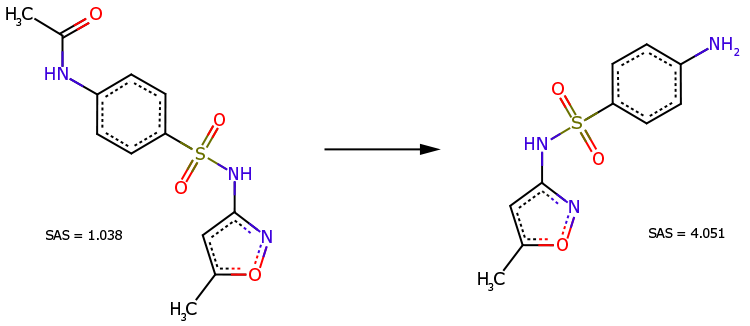
설파메톡사졸의 N-아세틸 유도체(그림 3, 왼쪽)는 이 약물의 직접 전구체입니다(그림 3, 오른쪽). 이 유도체는 화학 구조가 더 복잡하지만 합성이 더 쉬운 것으로 알려져 있습니다(SAS=1.038은 SAS=4.051보다 훨씬 작습니다).
사례 2
반면에 아드레날린의 N-Boc 유도체(그림 4, 왼쪽)는 아드레날린의 직접적인 전구체가 아닙니다(그림 4, 오른쪽). 일반적인 절차에서는 합성 경로 전체에서 아미노기를 보호할 필요가 없습니다. N-Boc 유도체는 합성 접근성 측면에서 더 복잡한 것으로 올바르게 인식됩니다(SAS=8.399가 SAS = 7.631보다 큼). 이는 아드레날린이 N-Boc 유도체 전구체라는 사실과 일치합니다.

사용자 데이터 흐름
SYNTHIA® SAS는 클라우드 호스팅 서비스로, RESTful API를 통해 각 고객이 사용할 수 있습니다. 수평적 확장이 가능하며 모든 고객에게 단일 API 진입점을 통해 고처리량을 제공합니다. 최종 사용자는 SMILES 형식의 분자 목록을 제공하면 SYNTHIA® SAS가 각 분자에 대한 점수를 반환합니다(그림 5). 이 서비스는 상태 비저장형이며 수요에 따라 확장할 수 있도록 설계되었습니다.

참고 자료
1. 조슈아 마이어스, 베네덱 파비안, 네이선 브라운, 새로운 분자 설계 및 생성 모델, 약물 발견의 오늘, 26, 2021, 2707-2715. DOI
2.SYNTHIA® 역합성 소프트웨어
3.Tomasz Klucznik 외, 컴퓨터로 계획하고 실험실에서 실행하는 다양한 의약 관련 표적의 효율성 있는 합성, Chem, 4, 2018,
522-532.DOI
4.Mikulak-Klucznik, B., 외. 복잡한 천연 제품의 합성에 대한 전산 계획, Nature, 588, 2020, 83-88. DOI
5.데이라이트 화학 정보 시스템, Inc.
6. 양, K., 외. 특성 예측을 위한 학습된 분자 표현 분석, 화학 정보 및 모델링 저널, 59, 2019, 3370-3388. DOI
7.Chemprop 오픈 소스 프로젝트
8.ChEMBL 데이터베이스
9.GDB 데이터베이스
