Biała księga:
Analiza obliczeniowa planowania syntetycznego: Przeszłość i przyszłość
Zaadaptowano z
Wang, Z., Zhang, W. i Liu, B. (2021), Computational Analysis of Synthetic Planning: Przeszłość i przyszłość. Chin. J. Chem., 39: 3127-3143. https://doi.org/10.1002/cjoc.202100273
Opublikowano dzięki uprzejmości Wiley.
Wspomagane komputerowo planowanie syntezy (CASP) może odgrywać znaczącą rolę w organizowaniu i wykorzystywaniu zalewu nowych reakcji chemicznych i reguł reakcji ekspertów do planowania nowatorskich i wysoce wydajnych syntez produktów naturalnych i kandydatów na leki. Niniejszy przegląd opisuje postępy w obliczeniowej analizie planowania syntezy od wczesnego etapu skupionego na programach opartych na regułach do uczenia maszynowego i ich połączonych możliwości.
Wprowadzenie
Chemicy wykorzystują analizę retrosyntetyczną do projektowania strategii syntetycznej dla związku docelowego. W skrócie, wykorzystują oni swoje doświadczenia w zrywaniu wiązań chemicznych w związku docelowym i kolejnych prekursorach w sposób iteracyjny.
Różne standardowe narzędzia (np. CML, SMILES, SMARTS, InChl i ECFP) tłumaczą reakcje chemiczne i cząsteczki na informacje czytelne dla maszyn. Bardziej zaawansowane algorytmy (np. sieci neuronowe, uczenie ze wzmocnieniem) rozszerzają przetwarzanie danych reakcji chemicznych.
Niniejszy przegląd obejmuje trzy kategorie CASP. Dwie kategorie wykorzystują logiczną dedukcję na podstawie intuicji i doświadczeń chemików: Algorytmy CASP oparte na ręcznie zakodowanych regułach lub na automatycznie wyodrębnionych regułach. Trzecia kategoria CASP wykorzystuje bazy danych reakcji chemicznych do szkolenia algorytmów uczenia maszynowego (ML).
Ogólna struktura systemu CASP
Typowy system CASP składa się z czterech modułów. Baza danych szablonów reakcji przechowuje znane reakcje z regułami zrywania wiązań. Moduł retrosyntetyczny dopasowuje znane reakcje w bazie danych szablonów do struktur cząsteczek wejściowych i zapewnia najbliższe dopasowanie do komercyjnie dostępnych prekursorów w sposób iteracyjny. Przewodnik po drzewach i moduł ewaluacyjny oceniają dopasowanie prekursorów do Ścieżek syntezy. Dostępna na rynku baza danych związków działa jako przystanek dla modułu retrosyntetycznego.
Ręcznie kodowane reguły w połączeniu z algorytmem logicznym
Reprezentatywne systemy CASP obejmują LHASA, SECS, IGOR, CHIRON i Chematica / SynthiaTM. Oba systemy CASP LHASA i SECS zawierały moduł komunikacyjny: połączony pad do pisania, aby chemicy mogli ocenić i wybrać najlepszą trasę z drzewa syntezy.
IGOR (Intermediate Generation of Organic Reactions) nie ograniczał analizy retrosyntetycznej do empirycznie wyprowadzonych reguł heurystycznych. IGOR obejmuje wszystkie cząsteczki uczestniczące w reakcji, wymaga obszernych obliczeń i może symulować tylko proste transformacje retrosyntetyczne.
CHIRON może dekodować złożoną stereochemię i funkcjonalność, które może skorelować z komercyjnie dostępnymi prekursorami wzbogaconymi o stereochemię. Wyszukuje prekursory o blisko spokrewnionych szkieletach, stereocentrach i grupach funkcyjnych z cząsteczką docelową.
Chematica (obecnie SynthiaTM) rozszerzyła sieć Chemii Organicznej (NOC) do około 10 milionów związków i ręcznie dodała informacje o zgodności i kontekście (np. warunki kanoniczne, nietolerancja grup funkcyjnych, regio- i stereoselektywność określonych reakcji) przy użyciu metody kodowania SMILES/SMART. Liczba ręcznie kodowanych reguł reakcji wzrosła do >100 000 w 2021 roku. Chematica/SynthiaTM zawiera inteligentną funkcję wyszukiwania, a funkcje punktacji chemicznej umożliwiają uzyskanie globalnie optymalnych wyników (np. chiralny prekursor do syntezy asymetrycznej).
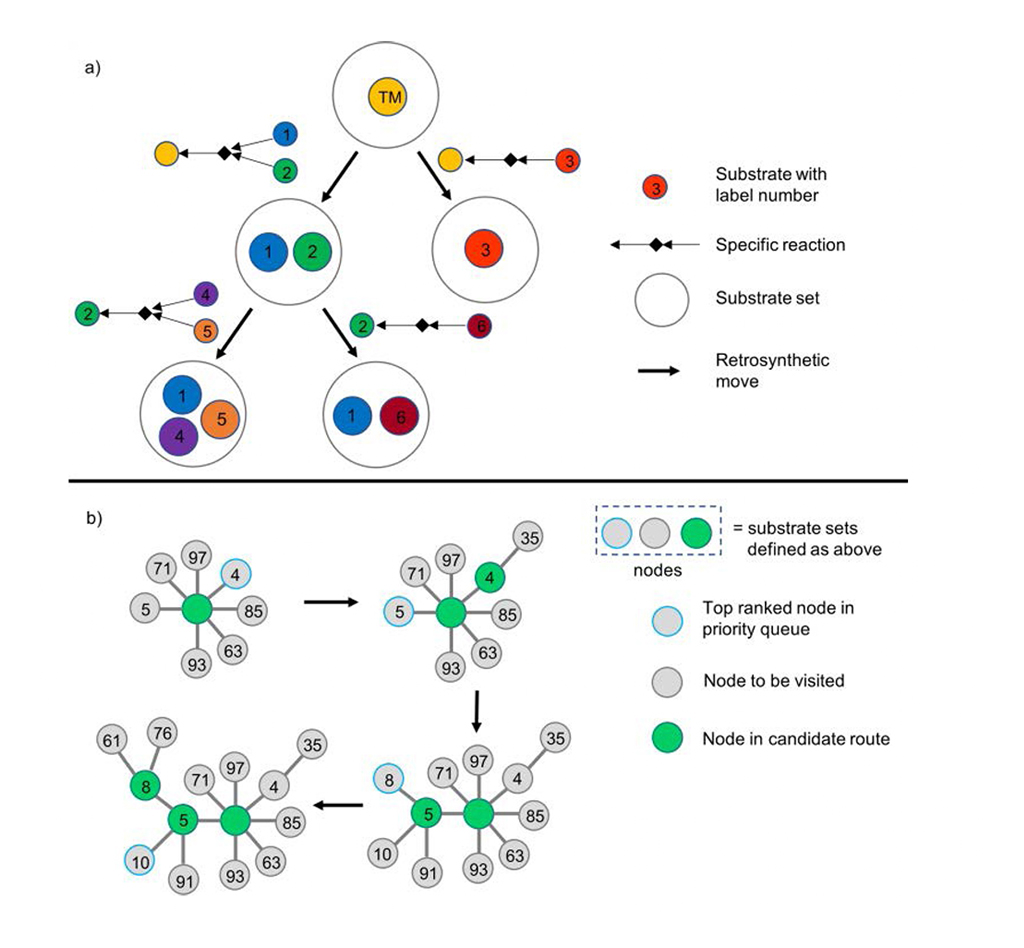
Chematica/SynthiaTM przedstawia drzewo syntetyczne w sposób dendrytyczny: każdy węzeł oznacza transformację retrosyntetyczną i powiązany z nią zestaw substratów (rys. 1a). Chematica/SynthiaTM przyspiesza proces analityczny dzięki kolejce priorytetowej dla węzłów o najniższej punktacji w algorytmie wyszukiwania (rys. 1b).
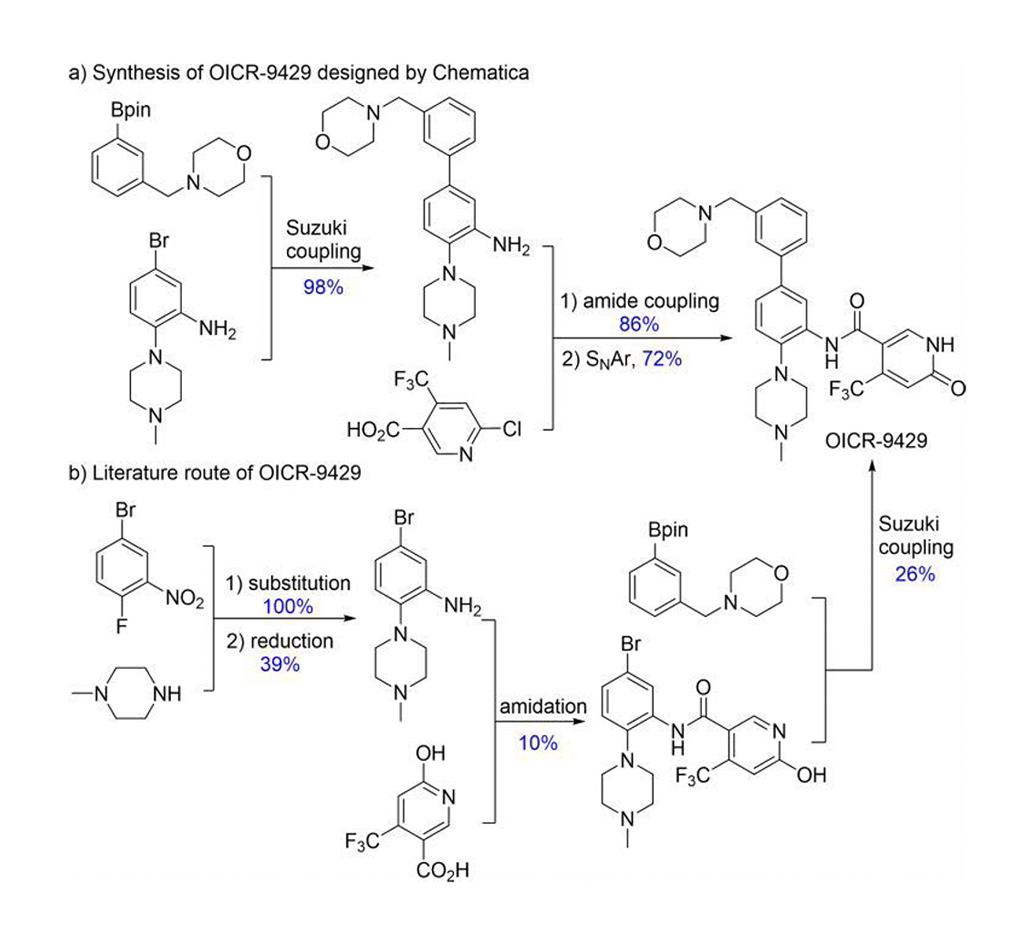
Chematica/SynthiaTM obejmuje różne metody mechaniki kwantowej i uczenia maszynowego (ML) w celu optymalizacji algorytmu wyszukiwania, funkcji punktacji i transformacji stereoselektywnych. Chematica/SynthiaTM zaprojektowała ścieżki syntezy dla ośmiu cząsteczek związanych z lekami i kilku złożonych produktów naturalnych. Ich syntezy zostały przeprowadzone eksperymentalnie. Program SynthiaTM zaprojektował bardziej wydajną drogę syntezy dla OICR-9429 (Rys. 2). Literatura donosiła o 1% wydajności OICR-9429; ale trasa SynthiaTM dała 60%. Co więcej, zaprojektowana przez SynthiaTM ścieżka syntezy uprościła oczyszczanie z czterech procedur chromatograficznych do jednej rekrystalizacji. W ten sposób Grzybowski i współpracownicy wyraźnie pokazują, że Chematica/SynthiaTM może rozwiązywać złożone problemy w chemii syntetycznej.
Ręczna ekstrakcja szablonów reakcji może poszerzyć informacje kontekstowe reakcji chemicznych i usprawnić analizy retrosyntetyczne. Wybór między automatyczną a ręczną ekstrakcją zależy od spójnego opisu zmiennych i pożądanych zastosowań.
Automatycznie wyodrębniane reguły w połączeniu z algorytmem logicznym
Automatyczne wyodrębnianie nowych reakcji chemicznych i szablonów codziennie może skutecznie utrzymywać bazy danych, ale może pomijać sąsiednie grupy funkcyjne i atomy.
SYNCHEM2 umożliwia zarówno wsteczne, jak i do przodu transformacje syntetyczne z alternatywnym kodowaniem. RETROSYN abstrahuje centrum reakcji i buduje korelację atomową między produktami i reagentami za pomocą specjalnego algorytmu różnicy grafów. RETROSYN wyszukuje i sortuje stopień dopasowania z priorytetem od wysokiego do niskiego, ale ignoruje stereochemię.
KOSP (Kowledge-base-Oriented System for Syntezy Planning) automatycznie wyodrębnia szablony reakcji, w tym grupy aktywujące/atomy w odległości trzech wiązań, aby wypełnić bazę wiedzy o reakcjach. Nowa wersja KOSP umożliwia analizę regio- i stereoselektywnej retrosyntezy, a aktualizacje rozszerzyły zawartość reakcji 10-krotnie.
ChemPlanner, następca ARChem, współpracuje na wyłączność z American Chemical Abstracts Service i Wiley w zakresie SciFinder, wysoce dostępnej bazy danych reakcji stworzonej przez naukowców. Nowa wersja ChemPlanner umożliwia analizę regio- i stereoselektywnej retrosyntezy.
ICSYNTH reprezentuje swoją bazę wiedzy o reakcjach w formie grafu. Użytkownicy mogą dołączać własne reguły chemiczne z poufnej bazy danych reakcji i dostosowywać ICSYNTH do różnych scenariuszy aplikacji, wybierając i edytując reguły chemiczne.
ASKCOS oblicza podobieństwo produktów reakcji do cząsteczki docelowej w celu opracowania planu retrosyntezy w sposób stopniowy. Moduły ASKCOS obejmują jednoetapową retrosyntezę, interaktywne planowanie ścieżek, tworzenie drzew i rekomendację kontekstową.
Automatycznie wyodrębnione reguły w połączeniu z algorytmem uczenia maszynowego
Algorytmy uczenia maszynowego są szkolone z wykorzystaniem baz danych reakcji chemicznych, w tym reagentów. Algorytmy uczenia ze wzmocnieniem stale współdziałają ze środowiskiem, które uczy je optymalnej strategii poprzez podejście kara-nagroda.
Program Bishop łączy opartą na regułach analizę retrosyntetyczną i uczenie ze wzmocnieniem. Sieć reakcji chemicznych zestawia półprodukty, łączy reagenty i produkty, a także posiada moduł uczenia ze wzmocnieniem w celu mapowania elastycznie zdefiniowanej, optymalnej ścieżki (ścieżek) reakcji z potencjalnymi filtrami kosztów, ogólnej Wydajności i/lub wpływu na środowisko.
3N-MCTS (algorytm Monte Carlo Tree Search) wykorzystuje sztuczne sieci neuronowe trenowane przez cyfrowe sekwencje produktów i odpowiednich prekursorów z literatury. Oparty na ANN system CASP reorganizuje określone wyuczone reguły reakcji, co upraszcza proces obliczeniowy. Każda runda MCTS składa się z selekcji, rozszerzenia, rozwinięcia i aktualizacji. Konieczne są ulepszenia w celu przewidywania stereoselektywności.
Model Seq2Seq z tłumaczeniem Simplified Molecular Input Line-Entry System (SMILES)
może przetwarzać ogromne zbiory danych i symulować reakcję z globalnym optymalnym wynikiem. AutoSynRoute ocenia ścieżki syntezy poprzez zastosowanie algorytmu MCTS z heurystycznymi funkcjami punktacji inspirowanymi przez Chematica/SynthiaTM. RXN wykorzystuje dwa retrosyntetyczne modele ML trenowane przez dwie bazy danych. RXN może przewidzieć odpowiednie warunki reakcji dla proponowanej ścieżki syntezy.
Wnioski
Kilka programów CASP stosuje heurystyczne reguły reakcji i reguły reakcji z literatury w swoich algorytmach dla chemii retrosyntetycznej z lub bez funkcji punktacji i ML (np. Chematica/SynthiaTM). Inne programy CASP opierają się na ML lub połączeniu ML z heurystycznymi regułami reakcji i/lub regułami chemicznymi opartymi na literaturze. Algorytmy te dostarczyły już nowych ścieżek syntezy, które poprawiły wydajność dla skomplikowanych cząsteczek. Dalsze ulepszenia mogą zapewnić nowe trasy syntezy złożonych związków z dodatkowymi ograniczeniami, takimi jak niższy koszt, mniejszy wpływ na środowisko i mniej niebezpiecznych odczynników lub rozpuszczalników.
