Nota aplikacyjna:
Ocena syntetyzowalności (SAS) firmy Synthia
Ocena syntetyzowalności (SAS) tysięcy wirtualnych cząsteczek w ciągu kilku minut.
Połącz swoje narzędzia cheminformatyczne z oprogramowaniem do retrosyntezy SYNTHIA® Oprogramowanie do retrosyntezy
Dostęp API
Dostęp API (Application Programming Interface) jest dostępny dla organizacji, które chciałyby połączyć inne narzędzia cheminformatyczne z oprogramowaniem SYNTHIA® w celu uzyskania niestandardowego doświadczenia.
Korzyści obejmują:
- Dostęp do pełnego interfejsu API retrosyntezy lub oceny syntetyzowalności (SAS)
- Wyświetlanie danych obok siebie w celu lepszego wglądu w wybór cząsteczek
- Tworzenie solidnych wizualizacji przy użyciu wielu źródeł danych
- Informowanie o wyborze cząsteczek przed etapem syntezy
- Analizowanie tysięcy ścieżek w ciągu kilku minut za pomocą interfejsu API SAS
Wykorzystaj moc oceny syntetyzowalności (SAS)
Zdolność do rozróżnienia między cząsteczkami "łatwymi do wykonania" i "trudnymi do wykonania" jest trudnym, ale bardzo przydatnym zadaniem, np. do ustalania priorytetów związków w wirtualnych potokach przesiewowych. Łącząc nowoczesny model głębokiego uczenia i dane zebrane za pomocą naszego renomowanego oprogramowania do planowania retrosyntetycznego, dostarczamy usługę SYNTHIA® Ocena syntetyzowalności (SAS), narzędzie mające zastosowanie do wysokoprzepustowego przetwarzania związków in-silico.
Obecnie chemia kombinatoryczna i modelowanie generatywne są wykorzystywane do konstruowania gigantycznych zbiorów danych związków [1]. Jednak rzeczywista synteza
wielu cząsteczek uzyskanych takimi metodami może stanowić wyzwanie. Aby rozwiązać ten problem, stosuje się syntetyczne miary dostępności w celu określenia wykonalności cząsteczek na jak najwcześniejszym etapie procesu odkrywania leków.
Usługa API SYNTHIA® SAS zapewnia prognozy takiej "złożoności molekularnej" pod względem liczby etapów syntezy z małych, dostępnych na rynku bloków konstrukcyjnych. Model uczenia maszynowego leżący u podstaw SAS został wstępnie wytrenowany na syntetycznych scenariuszach uzyskanych za pomocą algorytmów z SYNTHIA® Retrosynthetic Planning Tool [2], [3], [4]. Wreszcie, nasz hostowany w chmurze i posiadający certyfikat ISO-27001 produkt oferuje możliwość łatwego przetwarzania milionów cząsteczek dziennie i do tysiąca cząsteczek w pojedynczym zapytaniu, umożliwiając przewidywanie usług SYNTHIA® SAS do powszechniejszego stosowania w procesie projektowania leków.
Dane wejściowe/wyjściowe dla modelu SAS
Cząsteczki wejściowe muszą być dostarczone w szeroko stosowanym formacie tekstowym SMILES [5], a punkt końcowy API obsługuje żądania wsadowe. Wejściowe pliki SMILES składają się z pojedynczych fragmentów cząsteczek. Zwracana miara, tutaj zdefiniowana jako Ocena syntetyzowalności (SAS), jest pojedynczą liczbą zmiennoprzecinkową z zakresu 0-10, przypisaną dla każdej odpowiadającej cząsteczki wejściowej. Zwrócony wynik przybliża liczbę kroków potrzebnych do zsyntetyzowania cząsteczki przy użyciu komercyjnie dostępnych bloków konstrukcyjnych. Najniższe liczby (wartości bliskie 0) są zwracane dla związków chemicznych, które są przewidywane jako łatwe do wytworzenia (lub nawet mogą być dostępne komercyjnie). Wyższe liczby są zwracane, gdy model przewiduje więcej etapów syntezy w celu uzyskania żądanego związku. W przypadku wyników bliskich wartości maksymalnej (10) przewiduje się, że synteza będzie niezwykle złożona (wiele etapów reakcji) lub nawet niewykonalna, np. z powodu egzotycznych motywów strukturalnych w cząsteczce. Ogólnie rzecz biorąc, im niższy wynik, tym łatwiejsza powinna być synteza cząsteczki.
W przypadku, gdy niektóre cząsteczki w żądaniu są nieprawidłowe (np. hiperwartościowe, niekompletne pierścienie, nieprawidłowe protonowanie atomów aromatycznych, wielofragmentowe), żądanie będzie nadal przetwarzane. Wyniki dla takich wpisów będą zerowe, a odpowiednie komentarze zostaną zwrócone wraz ze strukturą odpowiedzi.
Charakterystyka modelu predykcyjnego
SYNTHIA® SAS v1.0 opiera się na regresorze, który obejmuje grafową konwolucyjną sieć neuronową (GCNN). Taka architektura pozwala na uczenie się wewnętrznej reprezentacji każdej cząsteczki poprzez operowanie na jej strukturze grafowej, a nie na wstępnie obliczonych deskryptorach molekularnych [6]. W szczególności model składa się z ukierunkowanej sieci neuronowej przekazującej wiadomości na poziomie wiązania (D-MPNN), po której następuje sieć neuronowa typu feedforward (FNN). Implementacja została zaadaptowana z otwartego projektu Chemprop [7].
Model uczenia maszynowego został wytrenowany przy użyciu wyników modułu automatycznej retrosyntezy SYNTHIA® jako wartości docelowej. Zastosowano wyspecjalizowany i znormalizowany wynik SYNTHIA®, aby odzwierciedlić liczbę kroków, np. nie penalizując nieselektywnych reakcji, ukrytą strategię ochrony, minimalny wkład ceny w wynik, a jako ustawienia wyszukiwania SYNTHIA® wykorzystano tylko małe bloki konstrukcyjne. Dodatkowo zastosowano funkcję wygładzania w celu lepszego budowania gradientu dla wysokich wyników, co miało na celu lepszą rozdzielczość trudnych do syntezy cząsteczek (patrz także rys. 1).
Dane wykorzystane do uczenia modeli uczenia maszynowego zawierają łącznie 33306 cząsteczek. Składa się on ze znanych cząsteczek (baza danych ChEMBL) [8] i kombinatorycznie wygenerowanych małych cząsteczek (GDB) [9]. Skład danych przed podziałem trening/test:
- podzbiór GDB: 16081, w tym:
- związki z 1-7 ciężkimi atomami (C, N, O, Cl, S): 7198
- związki z 8-9 ciężkimi atomami (C, N, O): 8883
- podzbiór ChEMBL: 17225, w tym:
- losowo wybrane syntetyczne małe związki: 15449
- losowo wybrane związki pochodzące z produktów naturalnych: 1776
Trening i ocena modelu uczenia maszynowego wymagały podzielenia danych na zestawy treningowe i testowe (zastosowano wspólny podział 80/20 trening/test). Ponadto,
wewnętrzny zestaw walidacyjny został wyodrębniony w stosunku 9:1 z zestawu treningowego i został wykorzystany do optymalizacji parametrów sieci.
Przewidywany wynik (model SYNTHIA® SAS) koreluje z wartością docelową opartą na wynikach SYNTHIA® z R2 = 0,726 i MAE = 1,1497. Wykres rozrzutu z dopasowaną linią i wykres pudełkowy pokazujący gęstość/rozkład punktów danych przedstawiono na Rys. 2.

Wyniki przewidywane za pomocą SYNTHIA® SAS są oparte na relacjach pobranych ze zbiorów danych (być może dość złożonych i niełatwych do uchwycenia). Należy to wziąć pod uwagę, gdy nowe cząsteczki są wyszukiwane za pośrednictwem interfejsu SYNTHIA® SAS-API. Mianowicie, wyniki dla cząsteczek, które nie są powiązane ze zbiorem testowym, mogą wypaść z tak zwanej domeny stosowalności, a zatem odpowiednie wyniki mogą nie być znaczące. Jest to typowe ograniczenie dla modeli opartych na danych, niemniej jednak zawsze dobrze jest pamiętać o takim ograniczeniu, aby uniknąć błędnej interpretacji uzyskanych wyników.
Studia przypadków
Przypadek 1
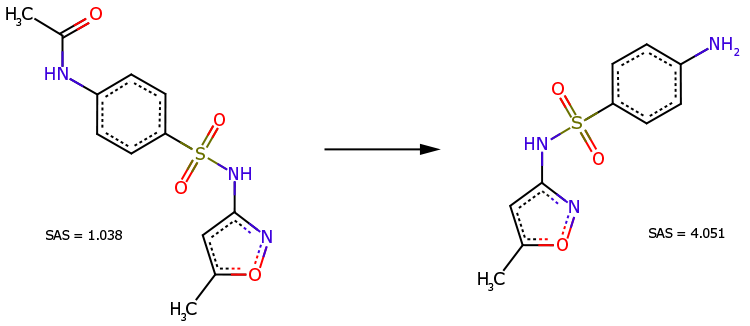
N-acetylowa pochodna sulfametoksazolu (Rys. 3, po lewej) jest bezpośrednim prekursorem tego leku (Rys. 3, po prawej). Pomimo bardziej złożonej struktury chemicznej, pochodna jest uznawana za łatwiejszą do syntezy (SAS=1,038 jest znacznie mniejszy niż SAS=4,051).
Przypadek 2
Z drugiej strony pochodna N-Boc adrenaliny (Rys. 4, po lewej) nie jest bezpośrednim prekursorem adrenaliny (Rys. 4, po prawej). W typowej procedurze nie ma potrzeby ochrony grupy aminowej na całej ścieżce syntezy. Pochodna N-Boc jest prawidłowo rozpoznawana jako bardziej złożona pod względem dostępności syntetycznej (SAS = 8,399 jest większy niż SAS = 7,631). Jest to zgodne z faktem, że adrenalina jest prekursorem swojej pochodnej N-Boc.

Przepływ danych użytkownika
SYNTHIA® API to usługa hostowana w chmurze, dostępna dla każdego klienta za pośrednictwem interfejsu API RESTful. Jest skalowalna horyzontalnie i zapewnia wysoką przepustowość poprzez pojedynczy punkt wejścia API dla wszystkich klientów. Użytkownik końcowy musi dostarczyć listę cząsteczek w formacie SMILES, a SYNTHIA® SAS zwraca wynik dla każdej z nich (rys. 5). Usługa jest bezstanowa i zaprojektowana do skalowania zgodnie z zapotrzebowaniem.

Odniesienia
1. Joshua Meyers, Benedek Fabian, Nathan Brown, De novo molecular design and generative models, Odkrywanie leków Today, 26, 2021, 2707-2715. DOI
2.SYNTHIA® Oprogramowanie do retrosyntezy
3.Tomasz Klucznik, et al., Wydajność syntezy różnorodnych, istotnych medycznie celów zaplanowanych komputerowo i wykonanych w laboratorium, Chem, 4, 2018,
522-532.DOI
4.Mikulak-Klucznik, B., et al. Computational planning of the synthesis of complex natural products, Nature, 588, 2020, 83-88. DOI
5.Daylight Chemical Information Systems, Inc.
6. Yang, K., et al. Analyzing Learned Molecular Representations for Property Prediction, Journal of chemical information and modeling, 59, 2019, 3370-3388. DOI
7.Projekt open-source Chemprop
8.Baza danych ChEMBL
9.Baza danych GDB
