Anwendungshinweis:
Synthia Synthetische Zugänglichkeitsbewertung (SAS)
Bewerten Sie die synthetische Zugänglichkeit (SAS) von Tausenden von virtuellen Molekülen in wenigen Minuten
Verbinden Sie Ihre Cheminformatik-Tools mit der SYNTHIA® Retrosynthese-Software
API-Zugang
Der API-Zugang (Application Programming Interface) ist für Unternehmen verfügbar, die andere Cheminformatik-Tools mit SYNTHIA® verbinden möchten, um eine maßgeschneiderte Lösung zu erhalten.
Die Vorteile umfassen:
- Zugriff auf die vollständige Retrosynthese- oder Synthetic Accessibility Score (SAS) API
- Betrachten Sie Daten Seite an Seite, um einen besseren Einblick in die Molekülauswahl zu erhalten
- Erstellen Sie robuste Visualisierungen unter Verwendung mehrerer Datenquellen
- Informieren Sie sich über die Molekülauswahl vor dem Syntheseschritt
- Analysieren Sie Tausende von Pfaden in wenigen Minuten mit der SAS API
Nutzen Sie die Leistungsfähigkeit des Synthetic Accessibility Score (SAS)
Die Fähigkeit, zwischen "leicht herstellbaren" und "schwer herstellbaren" Molekülen zu unterscheiden, ist eine schwierige, aber sehr nützliche Aufgabe, z. B. für die Priorisierung von Verbindungen in virtuellen Screening-Pipelines. Durch die Kombination des modernen Deep-Learning-Modells und der mit unserer renommierten Software für die retrosynthetische Planung gesammelten Daten liefern wir den SYNTHIA® Synthetic Accessibility Score (SAS) Service, ein Tool, das für die Verarbeitung von In-silico-Verbindungen im Hochdurchsatz geeignet ist.
Gegenwärtig werden die kombinatorische Chemie und die generative Modellierung für die Erstellung gigantischer Substanzdatensätze eingesetzt [1]. Die tatsächliche Synthese von
vielen Molekülen, die mit solchen Methoden erhalten wurden, kann jedoch eine Herausforderung darstellen. Um dieses Problem zu lösen, werden synthetische Zugänglichkeitsmaße verwendet, um die Machbarkeit von Molekülen so früh wie möglich in der Pipeline der Arzneimittelentdeckung zu bestimmen.
Der SYNTHIA® SAS API-Dienst liefert die Vorhersagen für eine solche "molekulare Komplexität" in Bezug auf die Anzahl der synthetischen Schritte aus kleinen, kommerziell erhältlichen Bausteinen. Das maschinelle Lernmodell, das SAS zugrunde liegt, wurde anhand synthetischer Szenarien trainiert, die mit Algorithmen des SYNTHIA® Retrosynthetic Planning Tool [2], [3], [4] gewonnen wurden. Schließlich bietet unser in der Cloud gehostetes und ISO-27001-zertifiziertes Produkt die Möglichkeit, täglich Millionen von Molekülen und bis zu tausend Moleküle in einer einzigen Abfrage zu verarbeiten, so dass die SYNTHIA® SAS-Servicevorhersage im Prozess des Wirkstoffdesigns immer häufiger eingesetzt werden kann.
Eingabe/Ausgabe für SAS-Modell
Die Eingabemoleküle müssen im weit verbreiteten SMILES-Textformat [5] bereitgestellt werden, und der API-Endpunkt unterstützt Batch-Anfragen. Die SMILES-Eingabe besteht aus einzelnen Molekülfragmenten. Das zurückgegebene Maß, hier definiert als Synthetic Accessibility Score (SAS), ist eine einzelne Fließkommazahl aus dem Bereich 0-10, die für jedes entsprechende Eingabemolekül vergeben wird. Der zurückgegebene Wert gibt an, wie viele Schritte zur Synthese des Moleküls unter Verwendung handelsüblicher Bausteine erforderlich sind. Die niedrigsten Zahlen (Werte nahe bei 0) werden für Chemikalien zurückgegeben, deren Herstellung als einfach vorhergesagt wird (oder die sogar kommerziell verfügbar sein können). Die höheren Zahlen werden zurückgegeben, wenn das Modell mehr synthetische Schritte vorhersagt, um die gewünschte Verbindung zu erhalten. Bei Werten nahe dem Maximalwert (10) wird die Synthese entweder als extrem komplex (viele Reaktionsschritte) oder sogar als undurchführbar vorhergesagt, z. B. aufgrund exotischer Strukturmotive im Molekül. Im Allgemeinen gilt: Je niedriger die Punktzahl, desto einfacher sollte die Synthese des Moleküls sein.
Falls einige der Moleküle in der Anfrage ungültig sind (z. B. hypervalent, unvollständige Ringe, unsachgemäße Protonierung aromatischer Atome, Mehrfachfragmente), wird die Anfrage dennoch bearbeitet. Die Punktzahlen für solche Einträge sind Null und entsprechende Kommentare werden in der Antwortstruktur mit angegeben.
Merkmale des Vorhersagemodells
SYNTHIA® SAS v1.0 basiert auf einem Regressor, der ein Graph Convolutional Neural Network (GCNN) enthält. Diese Architektur ermöglicht das Erlernen einer internen Repräsentation jedes Moleküls, indem sie mit seiner Graphenstruktur und nicht mit zuvor berechneten molekularen Deskriptoren arbeitet [6]. Insbesondere besteht das Modell aus einem gerichteten neuronalen Netzwerk auf Bindungsebene (D-MPNN), gefolgt von einem neuronalen Netzwerk mit Vorwärtssteuerung (FNN). Die Implementierung wurde vom Open-Source-Projekt Chemprop [7] übernommen.
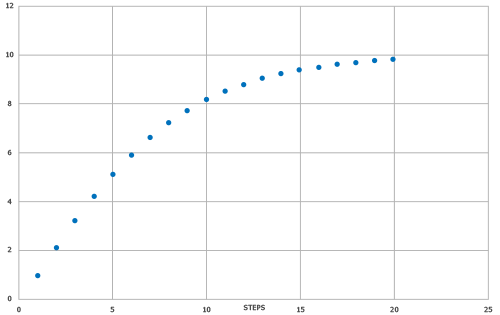
Das maschinelle Lernmodell wurde mit den Ergebnissen des automatischen Retrosynthesemoduls SYNTHIA® als Zielwert trainiert. Es wurde ein spezialisierter und normalisierter SYNTHIA®-Score verwendet, um die Anzahl der Schritte widerzuspiegeln, z.B. keine Bestrafung von nicht-selektiven Reaktionen, implizite Schutzstrategie, minimaler Preisbeitrag zum Score und nur kleine Bausteine wurden als SYNTHIA®-Sucheinstellungen verwendet. Zusätzlich wurde eine Glättungsfunktion angewandt, um einen besseren Gradienten für hohe Punktzahlen zu erzeugen, mit dem Ziel, schwer zu synthetisierende Moleküle besser aufzulösen (siehe auch Abb. 1).
Die für das Training der Modelle für maschinelles Lernen verwendeten Daten umfassen insgesamt 33306 Moleküle. Sie setzen sich aus bekannten Molekülen (ChEMBL-Datenbank) [8] und kombinatorisch generierten kleinen Molekülen (GDB) [9] zusammen. Die Zusammensetzung der Daten vor der Aufteilung in Training und Test:
- GDB-Teilmenge: 16081, darunter:
- Verbindungen mit 1-7 schweren Atomen (C, N, O, Cl, S): 7198
- Verbindungen mit 8-9 schweren Atomen (C, N, O): 8883
- ChEMBL-Teilmenge: 17225, darunter:
- zufällig ausgewählte kleine synthetische Verbindungen: 15449
- zufällig ausgewählte Verbindungen, die aus Naturprodukten gewonnen wurden: 1776
Für das Training und die Bewertung des maschinellen Lernmodells mussten die Daten in Trainings- und Testsätze aufgeteilt werden (es wurde ein üblicher 80/20-Train/Test-Split verwendet). Darüber hinaus wurde
der interne Validierungssatz im Verhältnis 9:1 aus dem Trainingssatz extrahiert und für die Optimierung der Netzwerkparameter verwendet.
Die vorhergesagte Punktzahl (SYNTHIA® SAS-Modell) korreliert mit dem Zielwert auf der Grundlage der SYNTHIA®-Scores mit R2 = 0,726 und MAE = 1,1497. Streudiagramm mit angepasster Linie und Boxplot, der die Dichte/Verteilung der Datenpunkte zeigt, sind in Abb. 2 dargestellt.

Die mit SYNTHIA® SAS vorhergesagten Ergebnisse basieren auf Beziehungen, die aus Datensätzen abgerufen werden (möglicherweise recht komplex und nicht einfach zu erfassen). Dies sollte berücksichtigt werden, wenn neue Moleküle über SYNTHIA® SAS-API abgefragt werden. Die Ergebnisse für Moleküle, die nicht mit dem Testsatz verwandt sind, könnten nämlich aus dem so genannten Anwendbarkeitsbereich herausfallen, so dass die entsprechenden Ergebnisse möglicherweise nicht aussagekräftig sind. Dies ist eine typische Einschränkung für datengesteuerte Modelle, dennoch ist es immer gut, sich diese Einschränkung vor Augen zu halten, um eine Fehlinterpretation der erzielten Ergebnisse zu vermeiden.
Fallstudien
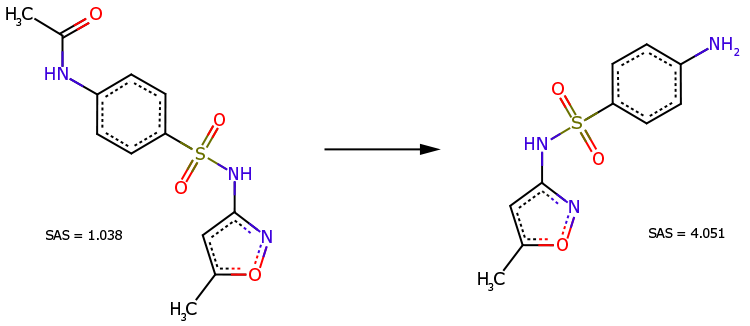
Fall 1
Das N-Acetylderivat von Sulfamethoxazol (Abb. 3, links) ist ein direkter Vorläufer dieses Arzneimittels (Abb. 3, rechts). Trotz der komplexeren chemischen Struktur gilt das Derivat als einfacher zu synthetisieren (SAS=1,038 ist viel kleiner als SAS=4,051).
Fall 2
Andererseits ist das N-Boc-Derivat von Adrenalin (Abb. 4, links) kein direkter Vorläufer von Adrenalin (Abb. 4, rechts). Bei einem typischen Verfahren ist es nicht erforderlich, die Aminogruppe während des gesamten Syntheseweges zu schützen. Das N-Boc-Derivat wird in Bezug auf die synthetische Zugänglichkeit korrekt als komplexer eingestuft (SAS = 8,399 ist größer als SAS = 7,631). Dies steht im Einklang mit der Tatsache, dass Adrenalin ein Vorläufer seines N-Boc-Derivats ist.

Benutzer-Datenfluss
SYNTHIA® SAS ist ein in der Cloud gehosteter Dienst, der für jeden Kunden über eine RESTful API verfügbar ist. Er ist horizontal skalierbar und bietet einen hohen Durchsatz über einen einzigen API-Einstiegspunkt für alle Kunden. Der Endnutzer muss eine Liste von Molekülen im SMILES-Format bereitstellen, und SYNTHIA® SAS gibt für jedes dieser Moleküle eine Bewertung zurück (Abb. 5). Der Dienst ist zustandslos und kann je nach Bedarf skaliert werden.

Referenzen
1. Joshua Meyers, Benedek Fabian, Nathan Brown, De novo molecular design and generative models, Drug Discovery Today, 26, 2021, 2707-2715. DOI
2.SYNTHIA® Retrosynthese-Software
3.Tomasz Klucznik, et al., Efficient Syntheses of Diverse, Medicinally Relevant Targets Planned by Computer and Executed in the Laboratory, Chem, 4, 2018,
522-532.DOI
4. Mikulak-Klucznik, B., et al. Computational planning of the synthesis of complex natural products, Nature, 588, 2020, 83-88. DOI
5.Daylight Chemical Information Systems, Inc.
6. Yang, K., et al. Analyzing Learned Molecular Representations for Property Prediction, Journal of chemical information and modeling, 59, 2019, 3370-3388. DOI
7.Chemprop Open-Source-Projekt
8.ChEMBL-Datenbank
9.GDB-Datenbank
