White Paper:
Computergestützte Analyse der synthetischen Planung: Vergangenheit und Zukunft
Angepasst von
Wang, Z., Zhang, W. und Liu, B. (2021), Computational Analysis of Synthetic Planning: Past and Future. Chin. J. Chem., 39: 3127-3143. https://doi.org/10.1002/cjoc.202100273
Veröffentlicht mit freundlicher Genehmigung von Wiley.
Die computergestützte Syntheseplanung (CASP) kann eine wichtige Rolle bei der Organisation und Nutzung der Flut neuartiger chemischer Reaktionen und fachkundiger Reaktionsregeln für die Planung neuartiger und hocheffizienter Synthesen von Naturprodukten und Arzneimittelkandidaten spielen. Diese Übersicht beschreibt den Fortschritt in der computergestützten Analyse der Syntheseplanung von der frühen Phase, die sich auf regelbasierte Programme konzentrierte, bis hin zum maschinellen Lernen und ihrer kombinierten Fähigkeit.
Einführung
Chemiker nutzen die retrosynthetische Analyse, um eine Synthesestrategie für eine Zielverbindung zu entwerfen. Kurz gesagt, sie nutzen ihre Erfahrungen beim Aufbrechen chemischer Bindungen in der Zielverbindung und den nachfolgenden Vorläufern in einer iterativen Weise.
Verschiedene standardisierte Tools (z. B. CML, SMILES, SMARTS, InChl und ECFP) übersetzen chemische Reaktionen und Moleküle in maschinenlesbare Informationen. Fortgeschrittenere Algorithmen (z. B. neuronale Netze, Verstärkungslernen) erweitern die Datenverarbeitung chemischer Reaktionen.
Diese Übersicht behandelt drei Kategorien von CASP. Zwei Kategorien nutzen logische Schlussfolgerungen aus den Intuitionen und Erfahrungen der Chemiker: CASP-Algorithmen, die auf handkodierten Regeln oder auf automatisch extrahierten Regeln basieren. Die dritte CASP-Kategorie verwendet chemische Reaktionsdatenbanken für das Training von Algorithmen für maschinelles Lernen (ML).
Allgemeiner Aufbau eines CASP-Systems
Ein typisches CASP-System besteht aus vier Modulen. Die Reaktionsvorlagendatenbank speichert bekannte Reaktionen mit Regeln für den Bindungsbruch. Das Retrosynthesemodul gleicht bekannte Reaktionen in der Schablonendatenbank mit Strukturen von Eingabemolekülen ab und liefert iterativ die beste Übereinstimmung mit kommerziell erhältlichen Vorläufern. Der Baumführer und das Bewertungsmodul bewerten die Übereinstimmung der Vorläuferkandidaten mit den Syntheserouten. Die Datenbank der kommerziell erhältlichen Verbindungen dient als Zwischenstation für das retrosynthetische Modul.
Handkodierte Regeln in Kombination mit logischen Algorithmen
Zu den repräsentativen CASP-Systemen gehören LHASA, SECS, IGOR, CHIRON und Chematica/SynthiaTM. Sowohl das LHASA- als auch das SECS-CASP-System enthielten ein Kommunikationsmodul: eine Schnittstelle zum Schreibblock, so dass Chemiker die beste Route aus dem Synthesebaum bewerten und auswählen konnten.
IGOR (Intermediate Generation of Organic Reactions) beschränkte die retrosynthetische Analyse nicht auf empirisch abgeleitete heuristische Regeln. IGOR umfasst alle an einer Reaktion beteiligten Moleküle, erfordert umfangreiche Berechnungen und kann nur einfache retrosynthetische Umwandlungen simulieren.
CHIRON ist in der Lage, komplexe Stereochemie und Funktionalität zu entschlüsseln, die es mit kommerziell erhältlichen, stereochemisch angereicherten Vorläufern in Beziehung setzen kann. Es sucht nach Vorläufern mit eng verwandten Skeletten, Stereozentren und funktionellen Gruppen des Zielmoleküls.
Chematica (jetzt SynthiaTM) hat das Netzwerk der Organischen Chemie (NOC) auf ca. 10 Millionen Verbindungen erweitert und manuell Kompatibilitäts- und Kontextinformationen (z. B. kanonische Bedingungen, Intoleranz funktioneller Gruppen, Regio- und Stereoselektivität spezifischer Reaktionen) unter Verwendung der SMILES/SMART-Kodierungsmethode hinzugefügt. Die Zahl der handkodierten Reaktionsregeln stieg bis 2021 auf über 100.000. Chematica/SynthiaTM verfügt über eine intelligente Suchfunktion und chemische Bewertungsfunktionen, die global optimale Ergebnisse ermöglichen (z. B. chirale Vorstufen für asymmetrische Synthesen).
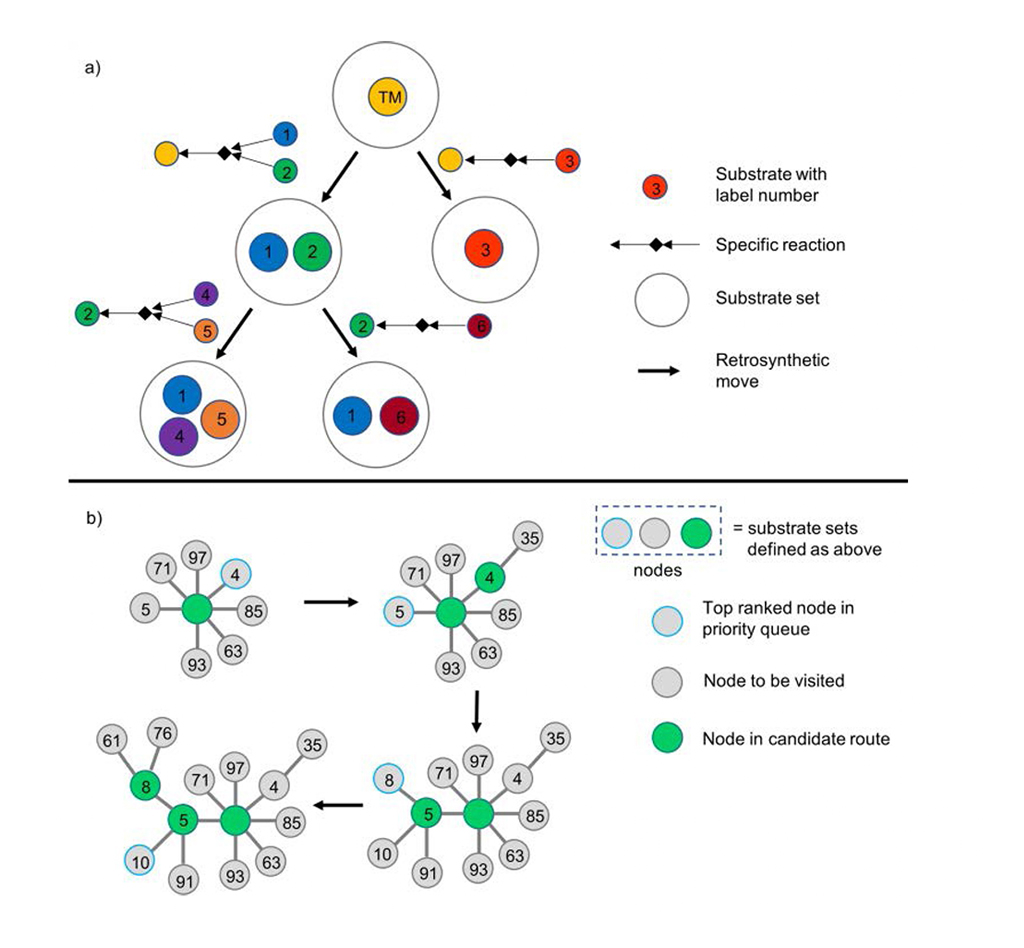
Chematica/SynthiaTM stellt den Synthesebaum dendritisch dar: Jeder Knoten bezeichnet die retrosynthetische Transformation und die dazugehörige Substratmenge (Abb. 1a). Chematica/SynthiaTM beschleunigt den analytischen Prozess mit einer Prioritätswarteschlange für die Knoten mit der niedrigsten Punktzahl im Suchalgorithmus (Abb. 1b).
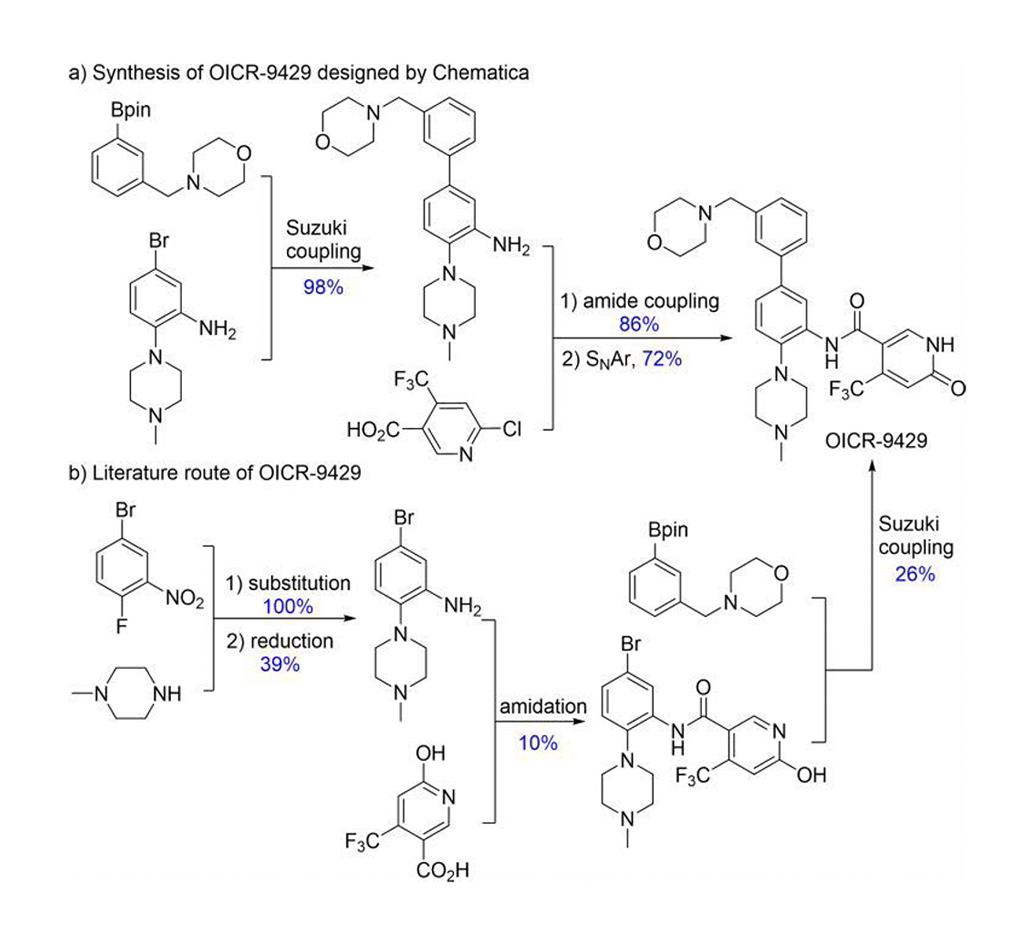
Chematica/SynthiaTM umfasst verschiedene Methoden der Quantenmechanik und des maschinellen Lernens (ML) zur Optimierung des Suchalgorithmus, der Bewertungsfunktionen und der stereoselektiven Transformationen. Chematica/SynthiaTM entwarf Syntheserouten für acht arzneimittelbezogene Moleküle und mehrere komplexe Naturprodukte. Ihre Synthesen wurden experimentell durchgeführt. Das Programm SynthiaTM entwarf einen effizienteren Syntheseweg für OICR-9429 (Abb. 2). In der Literatur wurde eine Ausbeute von 1 % für OICR-9429 angegeben, mit SynthiaTM wurden jedoch 60 % erreicht. Außerdem vereinfachte der von SynthiaTM entwickelte Syntheseweg die Reinigung von vier chromatographischen Verfahren auf eine Umkristallisation. Somit zeigen Grzybowski und seine Mitarbeiter deutlich, dass Chematica/SynthiaTM komplexe Probleme in der synthetischen Chemie lösen kann.
Die manuelle Extraktion von Reaktionsschablonen kann die Kontextinformationen von chemischen Reaktionen erweitern und retrosynthetische Analysen verbessern. Die Wahl zwischen automatischer und manueller Extraktion hängt von der konsistenten Beschreibung der Variablen und den gewünschten Anwendungen ab.
Automatisch extrahierte Regeln in Kombination mit einem logischen Algorithmus
Die tägliche automatische Extraktion neuer chemischer Reaktionen und Vorlagen kann die Datenbanken effizient verwalten, doch können dabei benachbarte funktionelle Gruppen und Atome übersehen werden.
SYNCHEM2 ermöglicht sowohl rückwärts als auch vorwärts gerichtete synthetische Transformationen mit alternativer Kodierung. RETROSYN abstrahiert das Reaktionszentrum und stellt mit einem speziellen Graphen-Differenz-Algorithmus eine atomare Korrelation zwischen Produkten und Reaktanden her. RETROSYN sucht und sortiert den Grad der Übereinstimmung mit einer hohen bis niedrigen Priorität, ignoriert aber die Stereochemie.
KOSP (Kowledge-base-Oriented System for Syntheseplanung) extrahiert automatisch Reaktionsvorlagen einschließlich aktivierender Gruppen/Atome innerhalb von drei Bindungsabständen, um die Reaction Knowledge Base aufzufüllen. Die neue KOSP-Version ermöglicht regio- und stereoselektive Retrosynthese-Analysen und Updates haben die Reaktionsinhalte um das 10-fache erweitert.
ChemPlanner, der Nachfolger von ARChem, kooperiert exklusiv mit dem American Chemical Abstracts Service und Wiley für SciFinder, eine leicht zugängliche Datenbank mit von Wissenschaftlern kuratierten Reaktionsinhalten. Die neue ChemPlanner-Version ermöglicht die Analyse von regio- und stereoselektiven Retrosynthesen.
ICSYNTH stellt seine Reaktionswissen-Datenbank in Form eines Graphen dar. Benutzer können eigene chemische Regeln aus der vertraulichen Reaktionsdatenbank einbinden und ICSYNTH für verschiedene Anwendungsszenarien anpassen, indem sie chemische Regeln auswählen und bearbeiten.
ASKCOS berechnet die Ähnlichkeit von Reaktionsprodukten mit dem Zielmolekül, um schrittweise einen retrosynthetischen Plan zu entwickeln. Zu den Modulen von ASKCOS gehören One-Step Retrosynthesis, Interactive Path Planning, Tree Builder und Context Recommendation.
Automatisch extrahierte Regeln in Kombination mit maschinellen Lernalgorithmen
ML-Algorithmen werden mit chemischen Reaktionsdatenbanken einschließlich Reaktanten trainiert. Die Algorithmen des Verstärkungslernens interagieren kontinuierlich mit der Umgebung, die ihnen die optimale Strategie über einen Strafe-Belohnung-Ansatz beibringt.
Das Bishop-Programm kombiniert regelbasierte retrosynthetische Analyse und Verstärkungslernen. Das chemische Reaktionsnetzwerk stellt die Zwischenprodukte zusammen, verbindet Reaktanten und Produkte und verfügt über ein Modul für verstärkendes Lernen, um einen flexibel definierten, optimalen Reaktionsweg bzw. -wege mit potenziellen Filtern für Kosten, Gesamteffizienz und/oder Umweltauswirkungen abzubilden.
3N-MCTS (Monte Carlo Tree Search-Algorithmus) verwendet künstliche neuronale Netze, die mit digitalen Sequenzen von Produkten und relevanten Vorläufern aus der Literatur trainiert werden. Das ANN-basierte CASP-System reorganisiert die spezifischen gelernten Reaktionsregeln, was den Berechnungsprozess vereinfacht. Jede MCTS-Runde besteht aus Selektion, Expansion, Rollout und Update. Verbesserungen sind erforderlich, um Stereoselektivitäten vorherzusagen.
Das Seq2Seq-Modell mit der SMILES-Übersetzung (Simplified Molecular Input Line-Entry System)
kann riesige Datensätze verarbeiten und eine Reaktion mit global optimalem Ergebnis simulieren. AutoSynRoute bewertet synthetische Wege durch Anwendung des MCTS-Algorithmus mit von Chematica/SynthiaTM inspirierten heuristischen Bewertungsfunktionen. RXN verwendet zwei retrosynthetische ML-Modelle, die mit zwei Datenbanken trainiert wurden. RXN kann geeignete Reaktionsbedingungen für den vorgeschlagenen Syntheseweg vorhersagen.
Schlussfolgerungen
Mehrere CASP-Programme verwenden heuristische Reaktionsregeln und Reaktionsregeln aus der Literatur in ihren Algorithmen für die retrosynthetische Chemie mit oder ohne Scoring-Funktionen und ML (z. B. Chematica/SynthiaTM). Andere CASP-Programme stützen sich auf ML oder die Kombination von ML mit heuristischen Reaktionsregeln und/oder chemischen Regeln aus der Literatur. Diese Algorithmen haben bereits neuartige Syntheserouten geliefert, die die Ausbeute bei komplizierten Molekülen verbessert haben. Weitere Verbesserungen können neuartige Syntheserouten für komplexe Verbindungen mit zusätzlichen Einschränkungen wie geringeren Kosten, geringerem ökologischen Fußabdruck und weniger gefährlichen Reagenzien oder Lösungsmitteln ermöglichen.
