Libro blanco:
Análisis computacional de la planificación sintética: Pasado y futuro
Adaptado de
Wang, Z., Zhang, W. y Liu, B. (2021), Computational Analysis of Synthetic Planning: Past and Future. Chin. J. Chem., 39: 3127-3143. https://doi.org/10.1002/cjoc.202100273
Publicado por cortesía de Wiley.
La planificación de síntesis asistida por ordenador (CASP) puede desempeñar un papel importante en la organización y el aprovechamiento de la avalancha de reacciones químicas novedosas y reglas de reacción expertas para la planificación de síntesis novedosas y altamente eficientes de productos naturales y candidatos a fármacos. Esta revisión describe los avances en el análisis computacional de la planificación sintética desde la fase inicial centrada en programas basados en reglas hasta el aprendizaje automático y su capacidad combinada.
Introducción
Los químicos utilizan el análisis retrosintético para diseñar una estrategia sintética para un compuesto diana. En pocas palabras, utilizan sus experiencias en la ruptura de enlaces químicos en el compuesto objetivo y los precursores posteriores de forma iterativa.
Diversas herramientas estandarizadas (por ejemplo, CML, SMILES, SMARTS, InChl y ECFP) traducen las reacciones químicas y las moléculas en información legible por máquina. Algoritmos más avanzados (por ejemplo, redes neuronales, aprendizaje por refuerzo) amplían el procesamiento de datos de reacciones químicas.
Esta revisión abarca tres categorías de CASP. Dos categorías utilizan la deducción lógica a partir de las intuiciones y experiencias de los químicos: Algoritmos CASP basados en reglas codificadas a mano o en reglas extraídas automáticamente. La tercera categoría CASP utiliza bases de datos de reacciones químicas para el entrenamiento de algoritmos de aprendizaje automático (ML).
Estructura general del sistema CASP
Un sistema CASP típico consta de cuatro módulos. La base de datos de plantillas de reacciones almacena reacciones conocidas con reglas de ruptura de enlaces. El módulo retrosintético alinea las reacciones conocidas de la base de datos de plantillas con las estructuras de las moléculas de entrada y proporciona la coincidencia más cercana con los precursores disponibles en el mercado de forma iterativa. La guía de árboles y el módulo de evaluación evalúan el ajuste de los precursores candidatos a las rutas sintéticas. La base de datos de compuestos disponibles en el mercado sirve de parada para el módulo retrosintético.
Reglas codificadas a mano combinadas con un algoritmo lógico
Algunos sistemas CASP representativos son LHASA, SECS, IGOR, CHIRON y Chematica/SynthiaTM. Tanto los sistemas CASP LHASA como SECS incluían un módulo de comunicación: un panel de escritura interconectado para que los químicos pudieran evaluar y seleccionar la mejor ruta del árbol sintético.
IGOR (Generación Intermedia de Reacciones Orgánicas) no restringía el análisis retrosintético a reglas heurísticas derivadas empíricamente. IGOR incluye todas las moléculas que participan en una reacción, requiere amplios cálculos y sólo puede simular transformaciones retrosintéticas sencillas.
CHIRON puede descodificar la estereoquímica y la funcionalidad complejas, que puede correlacionar con precursores enriquecidos en estereoquímica disponibles en el mercado. Busca precursores con esqueletos, estereocentros y grupos funcionales estrechamente relacionados con la molécula objetivo.
Chematica (ahora llamada SynthiaTM) ha ampliado la Red de Química orgánica (NOC) a unos 10 millones de compuestos y ha añadido manualmente información de compatibilidad y contexto (por ejemplo, condiciones canónicas, intolerancia de grupos funcionales, regioselectividad y estereoselectividad de reacciones específicas) utilizando el método de codificación SMILES/SMART. Sus reglas de reacción codificadas a mano aumentaron hasta >100.000 en 2021. Chematica/SynthiaTMincorpora una función de búsqueda inteligente y funciones de puntuación química que permiten obtener resultados globalmente óptimos (por ejemplo, precursor quiral para síntesis asimétrica).
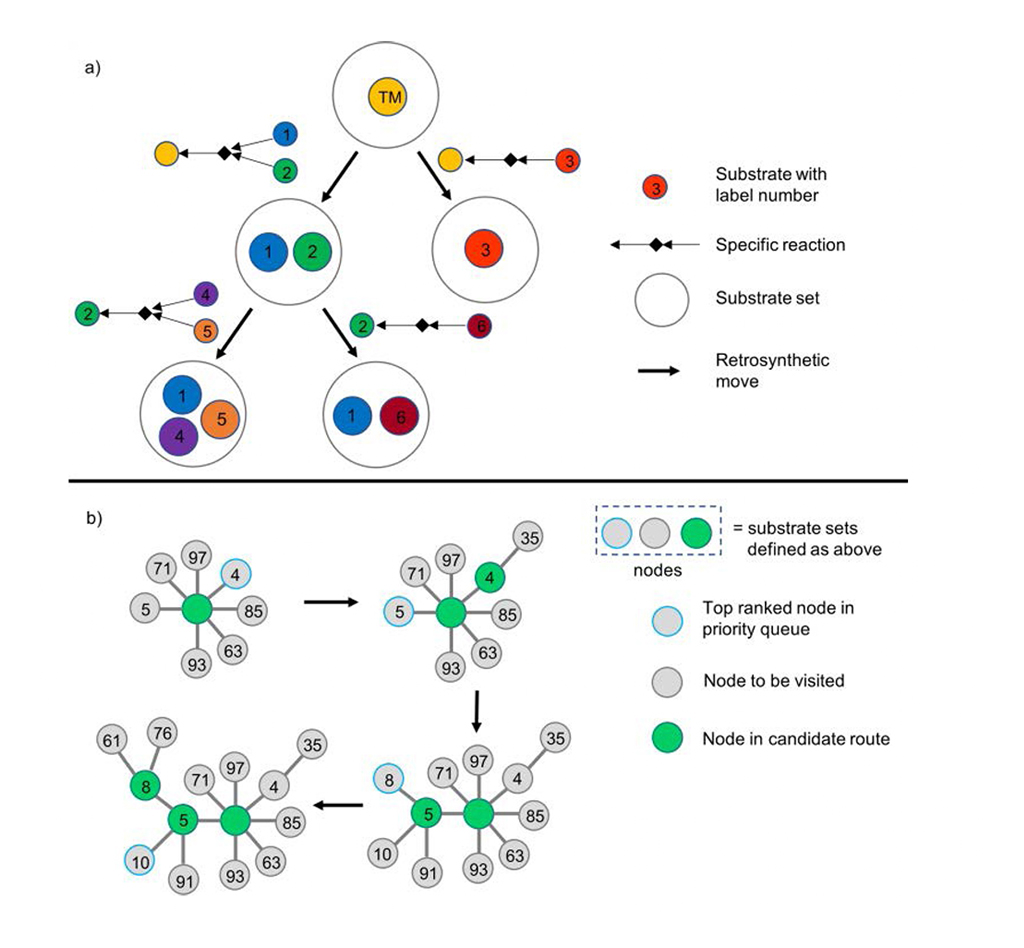
Chematica/SynthiaTM presenta el árbol sintético de forma dendrítica: cada nodo denota la transformación retrosintética y su conjunto de sustratos asociado (Fig. 1a). Chematica/SynthiaTM acelera el proceso analítico con una cola de prioridad para los nodos de menor puntuación en el algoritmo de búsqueda (Fig. 1b).
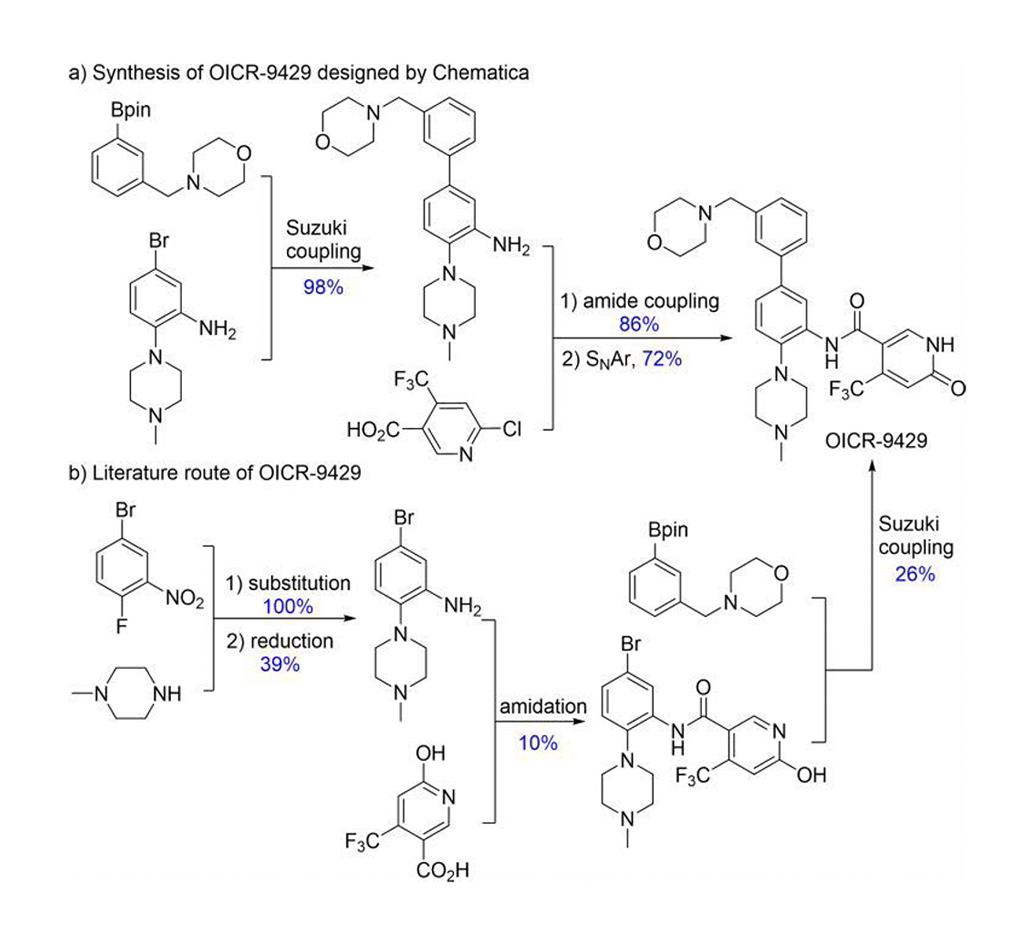
Chematica/SynthiaTM incluye varios métodos de mecánica cuántica y aprendizaje automático (ML) para optimizar el algoritmo de búsqueda, las funciones de puntuación y las transformaciones estereoselectivas. Chematica/SynthiaTM diseñó rutas sintéticas para ocho moléculas relacionadas con fármacos y varios productos naturales complejos. Sus síntesis se realizaron experimentalmente. El programa SynthiaTM diseñó una ruta sintética más eficiente para OICR-9429 (Fig. 2). Según la bibliografía, el rendimiento de OICR-9429 era del 1%, pero la ruta de SynthiaTM alcanzó el 60%. Además, la ruta sintética diseñada por SynthiaTM simplificó su purificación de cuatro procedimientos cromatográficos a una recristalización. Así, Grzybowski y colaboradores demuestran claramente que Chematica/SynthiaTM puede resolver problemas complejos en química sintética.
La extracción manual de plantillas de reacción puede ampliar la información de contexto de las reacciones químicas y mejorar los análisis retrosintéticos. La elección entre extracción automática y manual depende de la descripción coherente de las variables y de las aplicaciones deseadas.
Reglas extraídas automáticamente combinadas con un algoritmo lógico
La autoextracción diaria de nuevas reacciones químicas y plantillas puede mantener eficazmente las bases de datos, pero puede pasar por alto grupos funcionales y átomos adyacentes.
SYNCHEM2 permite transformaciones sintéticas hacia atrás y hacia delante con codificación alternativa. RETROSYN abstrae el centro de reacción y construye la correlación atómica entre productos y reactantes con un algoritmo especial de diferencia de grafos. RETROSYN busca y ordena el grado de coincidencia con una prioridad de alta a baja, pero ignora la estereoquímica.
KOSP (Kowledge-base-Oriented System for Planificación de la síntesis) extrae automáticamente plantillas de reacción que incluyen grupos/átomos activadores dentro de tres distancias de enlace para poblar la base de conocimientos de reacción. La nueva versión de KOSP permite el análisis de retrosíntesis regioselectiva y estereoselectiva, y las actualizaciones han multiplicado por 10 el contenido de las reacciones.
ChemPlanner, sucesor de ARChem, colabora en exclusiva con American Chemical Abstracts Service y Wiley para SciFinder, una base de datos muy accesible de contenidos de reacciones seleccionados por científicos. La nueva versión de ChemPlanner permite el análisis de retrosíntesis regioselectiva y estereoselectiva.
ICSYNTH representa su base de datos de conocimiento de reacciones en forma de base gráfica. Los usuarios pueden incluir reglas químicas propias de su base de datos confidencial de reacciones y adaptar ICSYNTH a diversos escenarios de aplicación seleccionando y editando reglas químicas.
ASKCOS calcula la similitud de los productos de reacción con la molécula objetivo para desarrollar un plan retrosintético de forma escalonada. Los módulos de ASKCOS incluyen la retrosíntesis en un solo paso, la planificación interactiva de rutas, el generador de árboles y la recomendación de contextos.
Reglas extraídas automáticamente combinadas con un algoritmo de aprendizaje automático
Los algoritmos de ML se entrenan con bases de datos de reacciones químicas que incluyen reactivos. Los algoritmos de aprendizaje por refuerzo interactúan continuamente con el entorno, que les enseña la estrategia óptima mediante un enfoque de penalización-recompensa.
El programa Bishop combina el análisis retrosintético basado en reglas y el aprendizaje por refuerzo. La Red de Reacción Química recopila los productos intermedios, conecta reactantes y productos, y dispone de un módulo de aprendizaje por refuerzo para trazar una(s) vía(s) de reacción óptima(s), definida(s) de forma flexible, con filtros potenciales de coste, eficiencia global y/o impacto medioambiental.
El algoritmo 3N-MCTS (Monte Carlo Tree Search algorithm) utiliza redes neuronales artificiales entrenadas mediante secuencias digitales de productos y precursores relevantes de la bibliografía. El sistema CASP basado en RNA reorganiza las reglas de reacción específicas aprendidas, lo que simplifica el proceso de cálculo. Cada ronda del MCTS consta de Selección, Expansión, Despliegue y Actualización. Se necesitan mejoras para predecir las estereoselectividades.
El modelo Seq2Seq con traducción Simplified Molecular Input Line-Entry System (SMILES)
puede procesar conjuntos de datos masivos y simular una reacción con un resultado óptimo global. AutoSynRoute evalúa rutas sintéticas aplicando el algoritmo MCTS con funciones de puntuación heurística inspiradas en Chematica/SynthiaTM. RXN utiliza dos modelos ML retrosintéticos entrenados mediante dos bases de datos. RXN puede predecir las condiciones de reacción adecuadas para la ruta sintética propuesta.
Conclusiones
Varios programas CASP aplican reglas de reacción heurísticas y reglas de reacción de la literatura en sus algoritmos para química retrosintética con o sin funciones de puntuación y ML (por ejemplo, Chematica/SynthiaTM) Otros programas CASP se basan en ML o en la combinación de ML con reglas de reacción heurísticas y/o reglas químicas basadas en la literatura. Estos algoritmos ya han proporcionado nuevas rutas sintéticas que mejoran el rendimiento de moléculas complicadas. Otras mejoras pueden proporcionar rutas sintéticas novedosas para compuestos complejos con restricciones adicionales como un menor coste, una menor huella medioambiental y menos reactivos o disolventes peligrosos.
