White Paper:
Analisi computazionale della pianificazione sintetica: Passato e futuro
Adattato da
Wang, Z., Zhang, W. e Liu, B. (2021), Analisi computazionale della pianificazione sintetica: Passato e futuro. Chin. J. Chem., 39: 3127-3143. https://doi.org/10.1002/cjoc.202100273
Pubblicato per gentile concessione di Wiley.
La pianificazione della sintesi assistita dal computer (CASP) può svolgere un ruolo significativo nell'organizzare e sfruttare la marea di nuove reazioni chimiche e le regole di reazione di esperti per pianificare sintesi inedite e altamente efficienti di prodotti naturali e candidati farmaci. Questa rassegna descrive i progressi nell'analisi computazionale della pianificazione sintetica, dalla fase iniziale incentrata sui programmi basati su regole all'apprendimento automatico e alla loro capacità combinata.
Introduzione
I chimici utilizzano l'analisi retrosintetica per progettare una strategia sintetica per un composto target. In breve, utilizzano la loro esperienza nella rottura dei legami chimici nel composto target e nei precursori successivi in modo iterativo.
Vari strumenti standardizzati (ad esempio, CML, SMILES, SMARTS, InChl ed ECFP) traducono le reazioni chimiche e le molecole in informazioni leggibili dalla macchina. Algoritmi più avanzati (ad esempio, reti neurali, apprendimento per rinforzo) ampliano l'elaborazione dei dati delle reazioni chimiche.
Questa rassegna copre tre categorie di CASP. Due categorie utilizzano la deduzione logica dalle intuizioni e dalle esperienze dei chimici: algoritmi CASP basati su regole codificate a mano o su regole estratte automaticamente. La terza categoria di CASP utilizza database di reazioni chimiche per l'addestramento di algoritmi di apprendimento automatico (ML).
Struttura generale del sistema CASP
Un tipico sistema CASP è composto da quattro moduli. Il database dei modelli di reazione memorizza le reazioni note con le regole di rottura dei legami. Il modulo di retrosintesi allinea le reazioni note nel database dei modelli con le strutture delle molecole in ingresso e fornisce la corrispondenza più vicina ai precursori disponibili in commercio in modo iterativo. La guida ad albero e il modulo di valutazione valutano l'adattamento dei precursori candidati alle vie sintetiche. Il database dei composti disponibili in commercio funge da tappa per il modulo di retrosintesi.
Regole codificate a mano combinate con un algoritmo logico
I sistemi CASP rappresentativi includono LHASA, SECS, IGOR, CHIRON e Chematica/SynthiaTM. Entrambi i sistemi CASP LHASA e SECS includevano un modulo di comunicazione: un blocco di scrittura interfacciato che consentiva ai chimici di valutare e selezionare la via migliore dall'albero sintetico.
IGOR (Intermediate Generation of Organic Reactions) non limitava l'analisi retrosintetica a regole euristiche derivate empiricamente. IGOR include tutte le molecole che partecipano a una reazione, richiede calcoli approfonditi e può simulare solo trasformazioni retrosintetiche semplici.
CHIRON è in grado di decodificare stereochimiche e funzionalità complesse, che può correlare a precursori arricchiti di stereochimica disponibili in commercio. Cerca precursori con scheletri, stereocentri e gruppi funzionali strettamente correlati alla molecola target.
Chematica (ora SynthiaTM) ha ampliato la Rete di Chimica Organica (NOC) a circa 10 milioni di composti e ha aggiunto manualmente informazioni sulla compatibilità e sul contesto (ad esempio, condizioni canoniche, intolleranza dei gruppi funzionali, regio- e stereoselettività di reazioni specifiche) utilizzando il metodo di codifica SMILES/SMART. Le regole di reazione codificate a mano sono aumentate a >100.000 nel 2021. Chematica/SynthiaTM ha incorporato una funzione di ricerca intelligente e funzioni di punteggio chimico che consentono di ottenere risultati globalmente ottimali (ad esempio, precursori chirali per sintesi asimmetriche).
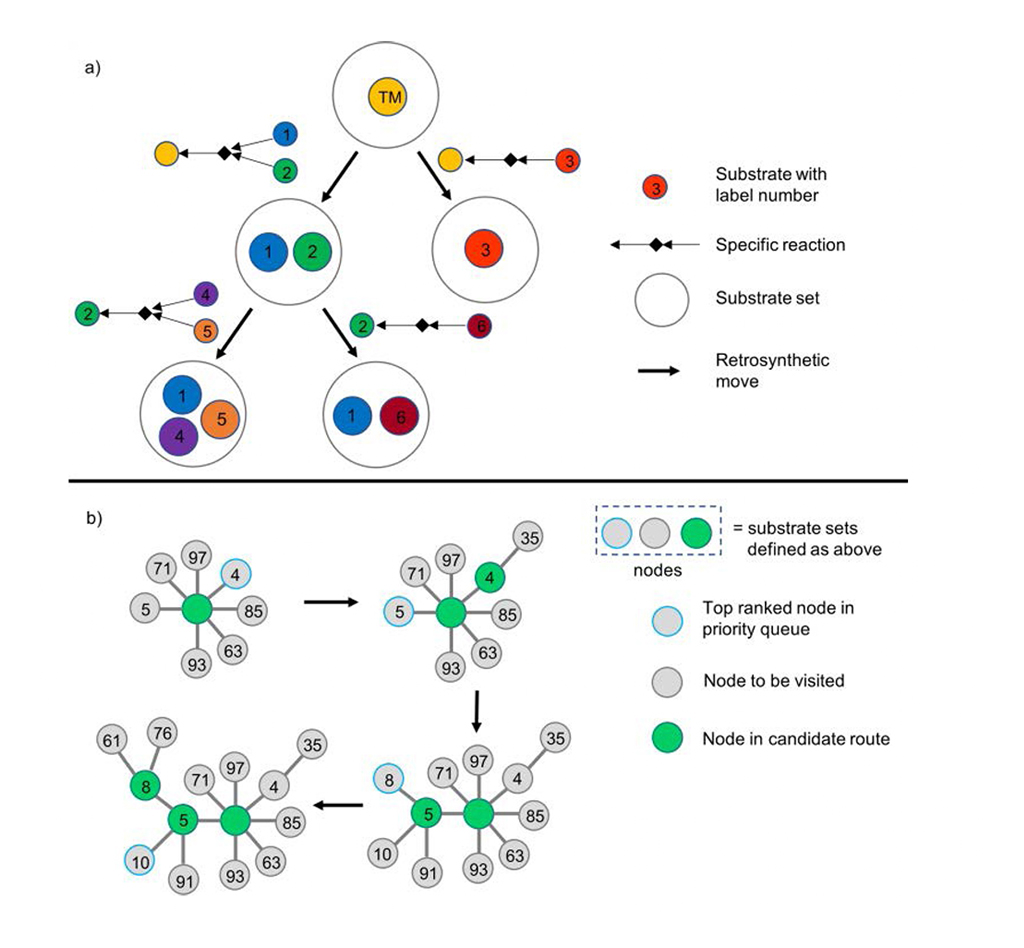
Chematica/SynthiaTM presenta l'albero sintetico in modo dendritico: ogni nodo denota la trasformazione retrosintetica e il set di substrati associato (Fig. 1a). Chematica/SynthiaTM accelera il processo analitico con una coda di priorità per i nodi con punteggio più basso nell'algoritmo di ricerca (Fig. 1b).
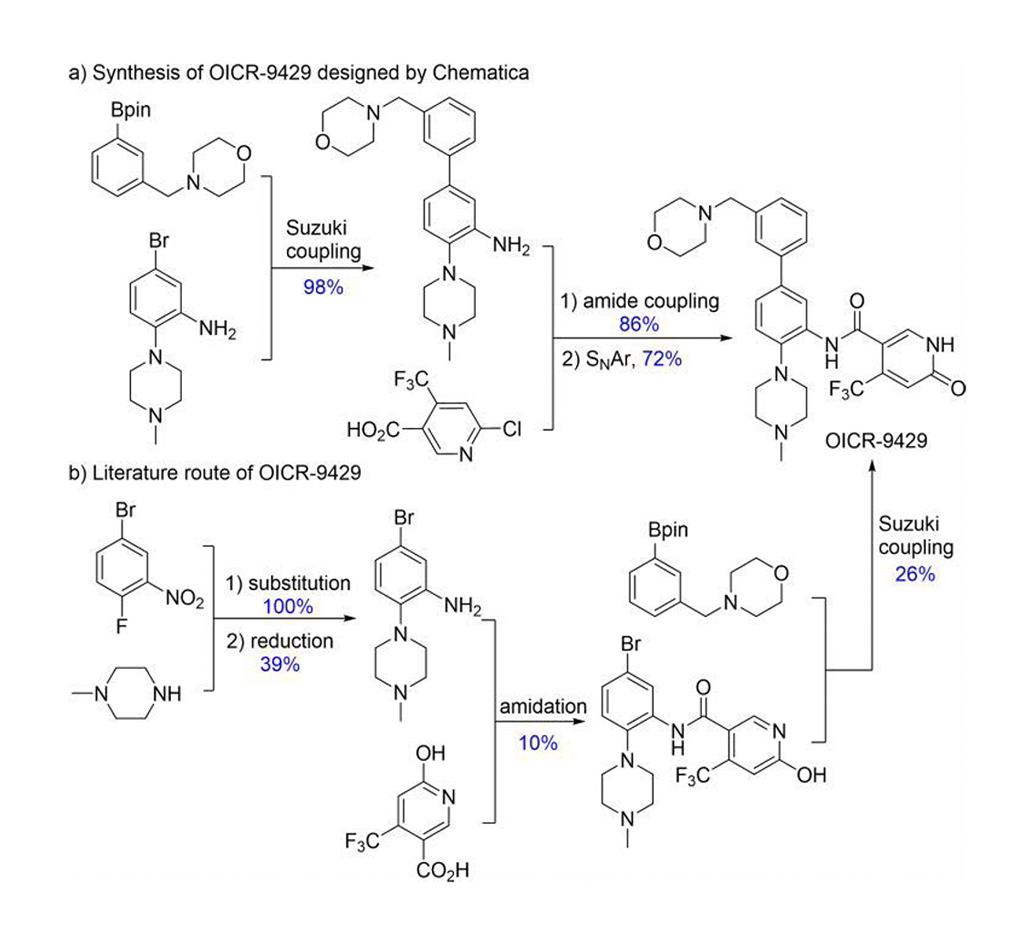
Chematica/SynthiaTM include vari metodi di meccanica quantistica e di apprendimento automatico (ML) per ottimizzare l'algoritmo di ricerca, le funzioni di punteggio e le trasformazioni stereoselettive. Chematica/SynthiaTM ha progettato vie sintetiche per otto molecole legate ai farmaci e per diversi prodotti naturali complessi. Le loro sintesi sono state realizzate sperimentalmente. Il programma SynthiaTM ha progettato una via sintetica più efficiente per OICR-9429 (Fig. 2). La letteratura riportava una resa dell'1% di OICR-9429, ma la via di SynthiaTM ha prodotto il 60%. Inoltre, la via sintetica progettata da SynthiaTM ha semplificato la purificazione, passando da quattro procedure cromatografiche a una ricristallizzazione. Pertanto, Grzybowski e collaboratori dimostrano chiaramente che Chematica/SynthiaTM può risolvere problemi complessi nella chimica sintetica.
L'estrazione manuale dei modelli di reazione può ampliare le informazioni sul contesto delle reazioni chimiche e migliorare le analisi di retrosintesi. La scelta tra estrazione automatica e manuale dipende dalla descrizione coerente delle variabili e dalle applicazioni desiderate.
Regole estratte automaticamente combinate con un algoritmo logico
L'estrazione automatica di nuove reazioni chimiche e di modelli ogni giorno può mantenere efficienti i database, ma può perdere gruppi funzionali e atomi adiacenti.
SYNCHEM2 consente trasformazioni sintetiche sia in avanti che all'indietro, con codifiche alternative. RETROSYN astrae il centro di reazione e costruisce correlazioni atomiche tra prodotti e reagenti con uno speciale algoritmo di differenza di grafo. RETROSYN cerca e ordina il grado di corrispondenza con una priorità da alta a bassa, ma ignora la stereochimica.
KOSP (Kowledge-base-Oriented System for Pianificazione della sintesi) estrae automaticamente modelli di reazione che includono gruppi/atomi attivanti entro tre distanze di legame per popolare la Reaction Knowledge Base. La nuova versione di KOSP consente l'analisi della retrosintesi regio- e stereoselettiva e gli aggiornamenti hanno ampliato il contenuto delle reazioni di 10 volte.
ChemPlanner, successore di ARChem, ha una collaborazione esclusiva con American Chemical Abstracts Service e Wiley per SciFinder, un database altamente accessibile di contenuti di reazione curati da scienziati. La nuova versione di ChemPlanner consente l'analisi della retrosintesi regio- e stereoselettiva.
ICSYNTH rappresenta il suo database di conoscenze sulle reazioni sotto forma di grafo. Gli utenti possono includere regole chimiche interne dal suo database di reazioni riservato e adattare ICSYNTH a vari scenari applicativi selezionando e modificando le regole chimiche.
ASKCOS calcola la somiglianza dei prodotti di reazione con la molecola target per sviluppare un piano di retrosintesi in modo graduale. I moduli di ASKCOS includono la retrosintesi in un solo passaggio, la pianificazione interattiva del percorso, il costruttore di alberi e la raccomandazione del contesto.
Regole estratte automaticamente combinate con algoritmi di apprendimento automatico
Gli algoritmi di ML sono addestrati con Basi di dati delle reazioni chimiche, compresi i reagenti. Gli algoritmi di apprendimento per rinforzo interagiscono continuamente con l'ambiente che insegna loro la strategia ottimale attraverso un approccio di penalità-ricompensa.
Il programma Bishop combina l'analisi retrosintetica basata su regole e l'apprendimento per rinforzo. Il Chemical Reaction Network compila gli intermedi, collega i reagenti e i prodotti e dispone di un modulo di apprendimento rinforzato per mappare uno o più percorsi di reazione ottimali, definiti in modo flessibile, con filtri potenziali per i costi, l'efficienza complessiva e/o l'impatto ambientale.
Il 3N-MCTS (algoritmo Monte Carlo Tree Search) utilizza reti neurali artificiali addestrate da sequenze digitali di prodotti e precursori rilevanti provenienti dalla letteratura. Il sistema CASP basato su RNA riorganizza le regole di reazione specifiche apprese, semplificando il processo di calcolo. Ogni ciclo MCTS è composto da Selezione, Espansione, Rilascio e Aggiornamento. Sono necessari miglioramenti per prevedere le stereoselettività.
Il modello Seq2Seq con il sistema di traduzione Simplified Molecular Input Line-Entry System (SMILES)
è in grado di elaborare enormi serie di dati e di simulare una reazione con un risultato ottimale globale. AutoSynRoute valuta le vie sintetiche applicando l'algoritmo MCTS con funzioni di punteggio euristiche ispirate a Chematica/SynthiaTM . RXN utilizza due modelli ML retrosintetici addestrati da due database. RXN è in grado di prevedere le condizioni di reazione adatte al percorso sintetico proposto.
Conclusioni
Diversi programmi CASP applicano regole di reazione euristiche e regole di reazione tratte dalla letteratura nei loro algoritmi per la chimica retrosintetica con o senza funzioni di scoring e ML (ad esempio, Chematica/SynthiaTM) Altri programmi CASP si basano su ML o sulla combinazione di ML con regole di reazione euristiche e/o regole chimiche basate sulla letteratura. Questi algoritmi hanno già fornito nuove vie sintetiche che hanno migliorato la resa di molecole complicate. Ulteriori miglioramenti possono fornire nuove vie sintetiche per composti complessi con vincoli aggiuntivi, come un costo inferiore, una minore impronta ambientale e un minor numero di reagenti o solventi pericolosi.
